VI Всероссийская научная молодежная школа-конференция
«Химия под знаком СИГМА: исследования, инновации, технологии»
18-20 мая 2020 г., Омск, Россия
http://conf.nsc.ru/sigma-6/ru
С 18 по 20 мая 2020 г. была успешно проведена VI Всероссийская научная молодежная школа-конференция «Химия под знаком СИГМА: исследования, инновации, технологии». Школа-конференция является традиционным мероприятием, проходящим один раз в два года в г. Омске. В 2020 в связи со сложившейся непростой эпидемиологической обстановкой по COVID-19 она впервые была организована в формате видеоконференции. Несмотря на непривычный формат, Школа-конференция вызвала большой интерес среди участников. Более 120 ученых из 10 городов России приняли активное участие в её работе. География мероприятия была весьма обширной. Самое большое количество участников было из научных и учебных учреждений Сибирского федерального округа: Омска, Новосибирска, Томска, Красноярска, Иркутска. Кроме того, доклады представили участники из Москвы, Санкт-Петербурга, Ростова на Дону, Казани.
Основными организаторами мероприятия выступили Институт катализа СО РАН и его Омский филиал – Центр новых химических технологий ИК СО РАН. Соорганизатором Школы-конференции был Омский государственный технический университет.
В рамках мероприятия были представлены лекции ведущих ученых-химиков и устные доклады молодых ученых, специалистов и преподавателей по современным направлениям фундаментальных и прикладных исследований в области химии и химической технологии. Научная тематика Школы включала два больших направления: «Кинетика и катализ» и «Синтез и исследование новых функциональных материалов». Каждый участник получил доступ ко всем представленным лекциям и докладам, имел возможность сделать устный доклад в режиме видеоконференцсвязи, а также получил эксклюзивный пакет участника.
Тезисы участников опубликованы в сборнике, который был выпущен в виде электронного издания сетевого распространения и размещен на официальном сайте Школы-конференции (раздел “Конференции” на сайте Института катализа СО РАН): http://catalysis.ru/resources/institute/Publishing/Report/2020/abstracts-sigma-2020.pdf.
Лучшие доклады отобраны для публикации в научных журналах. Среди изданий, обеспечивавших информационную поддержку Школы-конференции, были «Катализ в промышленности», «Журнал Сибирского федерального университета. Серия Химия», «AIP Conference Proceedings». Статьи выйдут в 2020 и 2021 гг.
Пленарные лекции Школы-конференции прочитали ведущие научные сотрудники институтов РАН и СО РАН из разных городов России. Затрагивались вопросы, связанные с созданием и использованием катализаторов для решения современных экологических проблем и установления детального механизма каталитического действия перспективных катализаторов и каталитических систем, а также применения и развития современных способов изучения катализаторов и каталитических реакций.
Открыл научную программу Школы-конференции д.х.н. Николай Юрьевич Адонин из Института катализа СО РАН (Новосибирск). Его доклад содержал сведения о катализаторах реакций органических соединений на основе фторированных арилборанов. В докладе были рассмотрены инструменты «грубой» и «тонкой» настройки для построения лигандного окружения атома бора, а также обсуждались три основных типа фторированных арилборанов: триарилбораны, диарилбориновые и арилбороновые кислоты. Представленные автором примеры были призваны продемонстрировать высокий потенциал фторированных арилборанов как эффективных гомогенных катализаторов для различного типа реакций органических соединений. Докладчик рассказал об использовании полифтортриарилборанов в каталитическом гидрировании, которое открывает возможность исключения переходных металлов из процессов синтеза биологически активных веществ, в частности, фармацевтических препаратов.
Выступление к.х.н. Андрея Валерьевича Бухтиярова (ИК СО РАН, Новосибирск) было посвящено применению метода РФЭС СИ для изучения гетерогенных каталитических систем. Был рассмотрен ряд серьезных методических трудностей, которые возникают в ходе изучения причин каталитической активности металлов в составе нанесенных катализаторов (проблемы “разрыва давлений” и “несоответствия материалов”), а также предложены пути их решения. Представлены примеры изучения ряда модельных нанесенных катализаторов, в которых металлические наночастицы нанесены на поверхность высокоориентированного пиролитического графита (ВОПГ) Ag/ВОПГ и Pd-Au/ВОПГ, в различных реакциях, в том числе в реакции окисления этилена на серебре до этиленоксида. Также было подробно рассказано о преимуществах биметаллических нанесенных катализаторов, связанных с тем, что введение второго компонента зачастую улучшает каталитические свойства монометаллических образцов. В качестве примера рассматривались Pd-Au катализаторы, проявляющие высокую каталитическую активность в целом ряде промышленно важных химических реакций. Были представлены результаты по синтезу и изучению сплавных Pd-Au наночастиц, нанесенных на поверхность ВОПГ, в реакциях окисления СО и гидрирования пропина. Следует отметить, что эксперименты по исследованию образцов в реакции окисления CO методом in situ РФЭС проводились в коллаборации Сибирских ученых с зарубежными исследователями на станции ISSIS Центра синхротронных исследований BESSY II (Берлин, Германия) в сотрудничестве с коллегами из Института Фрица-Габера общества Макса-Планка.
После перерыва в первый день работы Школы-конференции время для прочтения лекций было предоставлено учёным из Москвы. Свой доклад под названием «Прогресс в развитии универсальной теории катализа» академик Валентин Павлович Анаников (ИОХ РАН, Москва) начал с краткого экскурса в историю и увлекательно поведал об универсальной теории катализа, разработанной в 30-60 годы XX века выдающимся российским ученым в области катализа Алексеем Александровичем Баландиным. В докладе были рассмотрены современные механистические исследования каталитических процессов и актуальное состояние в области динамического катализа на металлических и углеродных центрах. Особое внимание было уделено вопросам взаимосвязи гомогенного и гетерогенного катализа и современным способам исследования каталитических реакций в твердофазных и жидкофазных системах
19 мая лекцию «Каталитические методы запасания и преобразования различных видов энергии» представил д.х.н. Денис Владимирович Козлов (Институт катализа СО РАН, Новосибирск). В докладе было рассказано непосредственно о путях трансформации и запасания энергии. Денис Владимирович отметил, что использование катализаторов позволяет снизить энергетические барьеры и повысить эффективность химических реакций, использующихся в процессах трансформации или запасания энергии. Чаще задачи по трансформации энергии возникают в области нетрадиционного катализа, связанного с искусственным фотосинтезом, водородной энергетикой, фотоэлектрохимическим преобразование солнечной энергии и др. Автор высказался о существующих на сегодняшний день подходах к реализации процессов трансформации и запасания различных видов энергии, а также способах их реализации, разработанных в Институте катализа СО РАН.
Выступление научного сотрудника Института катализа к.х.н. Ольги Александровны Булавченко было посвящено применению in situ рентгеновской дифракции для исследования оксидных и металл-оксидных катализаторов. Актуальность данной тематики обусловлена тем, что для разработки нового поколения катализаторов требуется не только развитие научно-практических основ их приготовления, но и понимание процессов, происходящих на всех стадиях синтеза и эксплуатации. Современные in situ методы анализа материалов позволяют следить за состоянием катализатора непосредственно в ходе реакции или в условиях, максимально приближенных к реальным. Докладчик рассказала о рентгенографических исследованиях методами in situ, которые позволяют изучить состояние веществ в различных внешних условиях. Было отмечено, что благодаря многочисленным методическим возможностям современной рентгенографии можно детально oхаракте¬ризовать происходящие под воздействием среды и температуры структурные изменения на атомном уровне. Применение in situ рентгеновской дифракции было рассмотрено на примере различных аспектов формирования Mn-Ce оксидных катализаторов.
В вечерней сессии второго дня работы Школы-конференции было представлено два доклада: д.х.н. Оксаны Павловны Таран из Института химии и химической технологии (г. Красноярск) и к.х.н. Романа Михайловича Мироненко из Центра новых химических технологий ИК СО РАН (г. Омск).
В лекции Оксаны Павловны Таран «Окисление воды до молекулярного кислорода одноэлектронными окислителями как краеугольный камень на пути создания каталитических систем для искусственного фотосинтеза и разложения воды на водород и кислород» затрагивались темы естественного и искусственного фотосинтеза, был дан обзор последних результатов, связанных с исследованием процесса окисления воды до молекулярного кислорода. Подробно было рассказано о коллоидных гидроксидных катализаторах, а также о гидроксидных катализаторах, стабилизированных цеолитами, которые применяются для данного процесса. Были всесторонне обсуждены кинетика и механизм процесса.
Доклад к.х.н. Романа Михайловича Мироненко назывался «Композиции на основе металлов платиновой группы и наноглобулярного углерода как катализаторы реакций органического синтеза». Автор отметил широкое применение катализаторов на основе углеродных материалов и нанесённых на них металлов платиновой группы (M/C, где M – в основном Ru, Rh, Pd и Pt). Он указал, что для синтеза катализаторов M/C используются специальные разновидности активного угля, получаемые из древесного и каменноугольного сырья, среди которых особое место занимает наноглобулярный углерод (НГУ), одной из разновидностей которого является производимый промышленностью технический углерод. В лекции были рассмотрены результаты, накопленные при исследовании композиций на основе металлов платиновой группы и НГУ. Основное внимание было уделено системам Pd/НГУ и их применению в качестве катализаторов для реакций гидрирования органических соединений. Помимо этого, в докладе были продемонстрированы перспективные направления дальнейших исследований каталитических композиций на основе НГУ и металлов платиновой группы.
20 мая научное заседание Школы-конференции открыл доклад «Новые катализаторы и каталитические процессы переработки биовозобновляемого сырья», сделанный д.х.н. Ольгой Владимировной Водянкиной из Томского государственного университета (Томск). Доклад был посвящен анализу существующих способов каталитической переработки основных «молекул–платформ», получаемых из переработанной биомассы, в ценные продукты, а также разработке новых подходов в области гетерогенного каталитического жидкофазного окисления полиолов, включая глицерин, 1,2-пропиленгликоль и 5-гидрокси-метилфурфурол. В лекции был представлен ряд перспективных подходов в области каталитической окислительной конверсии глицерина, пропиленгликоля и др. с использованием, в том числе, гибридных катализаторов на основе металлорганических координационных полимеров (МОКП) с иммобилизованными наночастицами металлов. Автор рассказала о новых экспериментальных результатах, полученных в области управления свойствами катализаторов типа МОКП UiO-66 путем введения модифицирующих заместителей различной природы в структуру лиганда – терефталевой кислоты, исследованных в процессе селективного окисления пропиленгликоля.
В своей лекции «Полифункциональный катализ в условиях термодинамического и кинетического контроля. Новые примеры» к.х.н. Евгений Анатольевич Булучевский (ЦНХТ ИК СО РАН, г. Омск) рассказал о разработке новых one-pot технологий, где в одном реакторе на полифункциональном катализаторе реализуется одновременно несколько стадий химического процесса. В качестве таких процессов были рассмотрены одностадийный синтез пропилена из этилена, получение пропилена метатезисом алкенов C5+ c этиленом, а также гидродеоксигенация масложирового сырья с получением низкозастывающих углеводородных компонентов моторных топлив. Евгений Анатольевич отметил, что процессы гидродеоксигенации кислородсодержащих молекул и изомеризации образующихся углеводородов протекают параллельно, при этом дезактивация центров различной природы на катализаторе происходит с разной скоростью. Также было показано, что быстрая дезактивация кислотных центров не оказывает существенного влияния на гидрирующую активность катализатора, однако приводит к снижению содержания изоалканов в продуктах.
Заключительными докладчиками Школы-конференкции были д.х.н. Б.Н. Кузнецов из Института химии и химической технологии (Красноярск) и к.х.н. Н.Н. Леонтьева из Центра новых химических технологий ИК СО РАН (Омск). В лекции профессора Бориса Николаевича Кузнецова «Процессы комплексной переработки (биорефайнери) возобновляемой древесной биомассы в ценные химические продукты» были представлены результаты последних исследований в области создания эффективных процессов каталитической переработки в востребованные химические вещества, функциональные полимеры и жидкие биотоплива возобновляемого лигноцеллюлозного сырья – древесной биомассы. Докладчик рассказал о перспективных направлениях исследований в создании принципиально новых технологий получения из лигноцеллюлозного сырья биотоплив и ценных химических продуктов, ориентированных на переработку всех основных компонентов лигноцеллюлозной биомассы (биорефайнери) с использованием катализаторов, нетоксичных реагентов и эффективных методов активации сырья.
Закрыл Школу-конференцию доклад к.х.н. Натальи Николаевны Леонтьевой из ЦНХТ ИК СО РАН (г. Омск) «Применение метода 1D моделирования рентгенограмм для исследования дефектных кристаллов». Она рассказала о рентгенографических методах, которые используются для исследования структуры слоистых дефектных кристаллов и позволяют уточнять типы образующихся дефектов. В докладе были представлены результаты применения метода 1D моделирования рентгенограмм к исследованию слоистых двойных гидроксидов (СДГ), имеющих гидроталькитоподобную структуру, а также оксидов на их основе. Как было продемонстрировано автором доклада, данный подход оказался наиболее информативным при изучении таких систем.
Несмотря на новый формат проведения Школы-конференции, все представленные доклады вызвали огромный интерес у слушателей, что отразилось в большом количестве вопросов и продуктивной дискуссии. На каждой секции были выбраны три лучших доклада молодых ученых. Их обладателями стали Валентин Сычев (ИХХТ КНЦ СО РАН), Максим Дубинин (ИК СО РАН) и Ольга Бугай (ЦНХТ ИК СО РАН) на секции «Кинетика и катализ», а также Шамиль Омаров (СпбГТУ), Максим Панафидин (ИК СО РАН) и Дмитрий Милордов (ИОФХ им. Арбузова КазНЦ РАН) на секции «Синтез и исследование функциональных материалов). Докладчики были отмечены почетными грамотами и получили памятные призы.
Материал подготовили:
Л.Н. Степанова, Н.Н. Леонтьева, М.А. Клюса, А.В. Лавренов
(Центр новых химических технологий ИК СО РАН, Омск;
Институт катализа СО РАН, Новосибирск)
3-я Всероссийская конференция
«Методы исследования состава и структуры функциональных материалов»
1-4 сентября 2020 г., Новосибирск, Россия
http://conf.nsc.ru/missfm-3/ru

1-4 сентября 2020 года в Новосибирском научном центре состоялась 3-я Всероссийская научная конференция «Методы исследования состава и структуры функциональных материалов». Конференция проводилась в режиме онлайн. Основными организаторами конференции являлись Институт катализа им. Г.К. Борескова СО РАН и Новосибирский государственный университет, со-организаторами стали Институт ядерной физики им. Г.И. Будкера СО РАН, Новосибирский Институт органической химии им. Н.Н. Ворожцова СО РАН, Институт неорганической химии им. А.В. Николаева СО РАН, Научный Совет по катализу.
Создание новых материалов и технологий – несомненный приоритет технологически развитых стран, стремящихся к высокому экономическому уровню. Материаловедением считают прикладную науку о взаимосвязи состава, строения и свойств материалов. Теоретической основой материаловедения являются соответствующие разделы физики и химии твердого тела, при этом разработка новых методов исследования строения, структуры, состава и физико-механических свойств материалов способствует дальнейшему развитию материаловедческих областей науки. Впечатляющими темпами развиваются технологии получения и методы характеризации широкого спектра функциональных материалов, в том числе магнитных и оптических материалов, полупроводников, сверхпроводников, химических источников тока, люминофоров, термохромных материалов, катализаторов, сорбентов, мембран, полимеров, а также наноматериалов, предназначенных для электроники, фотоники, информационных технологий, здравоохранения и экологии. Значение методов исследования состава и структуры функциональных материалов особенно велико как для целенаправленной оптимизации их свойств, так и для контроля качества при их производстве. Определение характеристик химического состава и структуры функциональных материалов является необходимым элементом исследований на всех этапах их синтеза, промышленного производства, при сертификации конечной продукции, а также в процессе эксплуатации.
Отличительной чертой конференции МИССФМ является ее междисциплинарный характер и приверженность обсуждению фундаментальных аспектов теории и практических подходов к методологии исследования состава и структуры широкого круга функциональных материалов.
Особое внимание на конференции МИССФМ-3 было уделено методам исследования наноструктурированных материалов с использованием синхротронного излучения, в том числе, в режимах in situ и operando (высокие температуры, давления, различные газовые и среды и т.д., имитирующие работу материала или устройства непосредственно в условиях его эксплуатации). Структурная сложность многих функциональных материалов требует применения методов исследования, выходящих далеко за рамки стандартного рентгеноструктурного анализа, а именно, установления структуры на разных уровнях организации вещества – от кристаллической структуры, локальной структуры атомного окружения и структуры поверхности до различных иерархических уровней нано- и микроструктуры. Разработка новых методов анализа применительно к структурным задачам стимулируется развитием экспериментальной техники (в частности, совершенствованием синхротронных источников, рентгенооптических элементов и детекторов, усложнением электронных микроскопов и систем пробоподготовки к ним) и определяется задачами экспресс-диагностики конденсированных сред, возможностью нового, синергетического использования большого объема дифракционных, спектральных, электронно-микроскопических и иных данных для решения актуальных задач современного материаловедения. Для всесторонней диагностики материалов используется широкий набор различных химических и физических методов, работают многочисленные коллективы исследователей. Поэтому стала очевидной необходимость критически рассмотреть и оценить возможности и области применения этих методов для решения междисциплинарных задач исследования всего спектра характеристик состава и структуры функциональных материалов и с учетом выявленных трендов сформировать общую программу развития данной области.
Конференция МИССФМ-3 собрала около 200 участников из 30 городов России, двух городов Германии и одного города Швеции. В рамках научной программы конференции было представлено 6 пленарных лекций, 6 ключевых лекций, 3 презентационных доклада, включая мастер-класс, 80 устных докладов, 33 устных доклада в Секции молодых ученых, 14 устных флэш-презентаций стендовой сессии, а также 39 стендовых докладов, размещенных на сайте конференции в формате PDF презентаций. Для обсуждения PDF презентаций стендовых докладов и проведения дискуссий по их содержанию был создан форум на основе сервиса Google Groups, который стал очень популярным среди участников.
Конференция была открыта двумя параллельными утренними сессиями Секции молодых ученых. Официальное открытие форума было назначено и состоялось после полудня для удобства участия в нем максимального количества исследователей – от Западной Европы до Дальнего Востока, с учетом того, что Новосибирск находится практически в центре часовых поясов. Европейские участники, желающие послушать представленные на утренней сессии доклады молодых ученых, имели доступ к их видео-записям.
 На официальном открытии выступили председатель Научного комитета конференции, директор Института катализа им. Г.К. Борескова СО РАН (ИК СО РАН), академик РАН, профессор Валерий Иванович Бухтияров; председатель конференции, заместитель директора ИК СО РАН, д.х.н. Олег Николаевич Мартьянов; заместитель председателя Научного комитета конференции, заместитель директора ИК СО РАН, член Программного комитета конференции, к.х.н. Алексей Анатольевич Ведягин; председатель Программного комитета конференции, заведующий лабораторией перспективных синхротронных методов исследования ИК СО РАН, д.ф.-м.н. Ян Витаутасович Зубавичус; заместитель председателя конферен-ции, заведующий лабораторией структурных методов ИК СО РАН, член Программного комитета конференции, д.ф.-м.н. Сергей Васильевич Цыбуля.
На официальном открытии выступили председатель Научного комитета конференции, директор Института катализа им. Г.К. Борескова СО РАН (ИК СО РАН), академик РАН, профессор Валерий Иванович Бухтияров; председатель конференции, заместитель директора ИК СО РАН, д.х.н. Олег Николаевич Мартьянов; заместитель председателя Научного комитета конференции, заместитель директора ИК СО РАН, член Программного комитета конференции, к.х.н. Алексей Анатольевич Ведягин; председатель Программного комитета конференции, заведующий лабораторией перспективных синхротронных методов исследования ИК СО РАН, д.ф.-м.н. Ян Витаутасович Зубавичус; заместитель председателя конферен-ции, заведующий лабораторией структурных методов ИК СО РАН, член Программного комитета конференции, д.ф.-м.н. Сергей Васильевич Цыбуля.
С теплым приветствием к участникам обратился Валерий Иванович Бухтияров, выразив свой интерес к новому формату мероприятия, а особенно к опыту проведения стендовой сессии и интерактивного вечернего фуршета, поскольку как организаторы, так и участники в осуществлении этих новшеств практически являлись первопроходцами. Переходя к научной составляющей конференции, Валерий Иванович отметил, что именно развитие физических методов и их профессиональное использование позволяют прийти к решению основных фундаментальных вопросов катализа. Институт катализа всегда активно участвовал в развитии физических методов, поэтому в свое время он стал организатором этой конференции. Задачей конференции МИССФМ-3 он обозначил возможность собрать вместе исследователей, развивающих разные физические методы, чтобы они могли определить тот набор адекватных методов, который позволил бы свести воедино данные об изучаемом объекте. Развитие физических методов определяется в том числе созданием синхротронных центров. Поскольку исследователи Института катализа активно используют существующие два центра в России, а также и гораздо более мощные международные центры, в 2018 году на встрече с В.В. Путиным Институт катализа вместе с Институтом ядерной физики им. Г.И. Будкера СО РАН выступил инициатором создания Центра коллективного пользования «Сибирский кольцевой источник фотонов» (СКИФ). В конечном итоге появился Указ Президента о развитии сети установок класса «MEGASCIENCE», куда попал и ЦКП «СКИФ». В заключение Валерий Иванович рассказал о характеристиках проектируемого источника фотонов, о планах и сроках осуществления проекта, о проделанных на сегодняшний день работах, достигнутых договоренностях и подписанных контрактах.
Олег Николаевич Мартьянов, также отметив необычный формат конференции, напомнил, что тенденция и логика прогресса современной науки свидетельствуют о том, что наиболее яркие результаты получаются в различных областях с использованием открывшихся экспериментальных возможностей. При этом он привел в пример последние работы, удостоенные нобелевских премий в области физики, биологии, химии. В развитие мыслей, высказанных Валерием Ивановичем Бухтияровым, Олег Николаевич подтвердил, что каким бы уникальным ни был тот или иной метод, на разных уровнях для решения научных задач требуется комплекс взаимодополняющих методов, которые позволяют найти место для новых экспериментальных возможностей по решению многофункциональных задач. Поэтому Центры коллективного пользования, которые используют настолько разные методы исследования, что их объединение на первый взгляд выглядит непонятным, получают конкурентное преимущество. Направления их деятельности повторяют тематику конференции МИССФМ-3, которая практически и является мостиком для разных методов исследований. Поэтому она очень востребована и вызывает большой интерес.
Алексей Анатольевич Ведягин рассмотрел организационные моменты предстоящей конференции, напомнив о некоторых ограничениях, которые накладывает разница в часовых поясах участников вследствие их географической удаленности друг от друга. Стремление ученых из разных географических точек принять участие в работе конференции, с одной стороны, очень радует и является ярким показателем интереса к форуму, но, с другой стороны, усложняет формирование расписания заседаний. Алексей Анатольевич подчеркнул, что организаторы сделали все возможное, чтобы создать удобную сетку для эффективного и комфортного участия в научных сессиях, обеспечив общий доступ к записям докладов в случае необходимости. Возглавляя работу команды научно-технических модераторов онлайн сервиса, он напомнил участникам правила использования платформы ZOOM при представлении докладов и участии в дискуссиях. Алексей Анатольевич, будучи руководителем Отдела материаловедения и функциональных материалов, выразил уверенность что на конференции будут представлены новые подходы к различным методам исследования функциональных свойств материалов, в том числе таких, которые ранее оставались невостребованными, поскольку их доступность определяет эффективность использования материалов.
Ян Витаутасович Зубавичус посвятил свое приветственное выступление основной официальной подтеме научной программы конференции – использованию синхротронного излучения. Акцен¬тирование этой тематики связано с тем, что ИК СО РАН в консорциуме с целым рядом сибирских институтов ведет очень крупный проект по созданию в Новосибирской области суперсовременного нового Центра синхротронного излучения поколения 4+. Это даст большой толчок развитию науки не только в Сибирском регионе, но и во всей России. Научное сообщество на данный момент должно определить, какие задачи будут решаться на синхротронном источнике, поскольку это будет междисциплинарный, многофункциональный исследовательский центр, призванный решать задачи в самых разных областях науки. Ян Витаутасович призвал всех участников знакомиться друг с другом, образовывать новые консорциумы, генерировать новые идеи и обмениваться ими, предлагать новые методы или их комбинации, чтобы расширять возможности современных подходов для решения самых разных исследовательских задач.
Сергей Васильевич Цыбуля познакомил аудиторию с некоторыми статистическими данными. Он отметил востребованность конференции МИССФМ-3, в основе которой лежит комплексность и междисциплинарность исследований. Сергей Васильевич анонсировал организацию стендовой сессии, призвал участников к активному диалогу на созданном в Google Groups форуме для обсуждения загруженных на сайт стендовых презентаций. Он также подчеркнул важность участия в конференции большого количества молодых ученых, аспирантов и студентов. Сергей Васильевич проинформировал аудиторию, что несколько кафедр Новосибирского государственного университета предпочли начать учебный год с того, чтобы направить студентов в первые дни сентября посещать онлайн сессии конференции вместо запланированных классов.
В своих приветствиях выступающие поблагодарили участников за проявление активности в такой непростой период времени, пожелали плодотворной работы, интересных докладов, продуктивных дискуссий, приобретения новых знаний, оптимизма, хорошего настроения.
 Пленарная сессия была представлена шестью известными специалистами в области физико-химии функциональных материалов, экспертами мирового уровня. Ее открыл доктор Андрей Шаворский, научный сотрудник лаборатории MAX IV – шведской национальной лаборатории исследований с использованием синхротронного излучения, лекцией “Scientific opportunities at MAX IV, the 4th generation synchrotron radiation source” (Исследовательские возможности источника синхротронного излучения 4-го поколения MAX IV). Доктор Шаворский представил информацию об истории создания лаборатории синхротронного излучения, дал краткое описание важных технических характеристик и концепций, лежащих в основе создания установки, привел несколько ярких примеров ее использования, основанных на уникальных свойствах излучения, генерируемого ускорителями MAX IV.
Пленарная сессия была представлена шестью известными специалистами в области физико-химии функциональных материалов, экспертами мирового уровня. Ее открыл доктор Андрей Шаворский, научный сотрудник лаборатории MAX IV – шведской национальной лаборатории исследований с использованием синхротронного излучения, лекцией “Scientific opportunities at MAX IV, the 4th generation synchrotron radiation source” (Исследовательские возможности источника синхротронного излучения 4-го поколения MAX IV). Доктор Шаворский представил информацию об истории создания лаборатории синхротронного излучения, дал краткое описание важных технических характеристик и концепций, лежащих в основе создания установки, привел несколько ярких примеров ее использования, основанных на уникальных свойствах излучения, генерируемого ускорителями MAX IV.
MAX IV – первый в мире дифракционно-ограниченный источник синхротронного излучения. Эта установка синхротрон-ного излучения является синхротроном нового, 4-го поколения, его запуск ознаменовал начало новой эры для рентгеновской науки. Открытие лаборатории синхротронного излучения MАХ IV состоялось в июне 2016 года, а четыре года спустя в нем уже было 10 исследовательских станций, еще 6 установок для пользователей сейчас находятся на разных этапах строительства или ввода в эксплуатацию. После полного завершения MAX IV будет иметь 26 рабочих станций, использующих излучение вставных устройств, размещенных на трех ускорителях: накопительные кольца на 1,5 ГэВ и 3 ГэВ и линейный ускоритель на 3 ГэВ. Благодаря успешной реализации инновационного дизайна магнитной структуры «7MBA» на накопительном кольце 3 ГэВ MAX IV удалось достичь рекордного в мире эмиттанса 300 пм·рад (или 200 пм·рад с установленными вставными устройствами). Это позволило достичь беспрецедентного уровня яркости и степени когерентности, что открыло новые возможности в широком спектре научных областей с многократно улучшенным пространственным, энергетическим и временным разрешением.
МАХ IV является самой современной установкой синхротронного излучения. Лаборатория располагает уникальным оборудованием, которое позволяет получать новые знания о молекулах и их структуре, открывает новые возможности для понимания свойств и структуры биологических систем, новых функциональных материалов, наноматериалов и композитов. Кроме того, интенсивные когерентные пучки, создаваемые ускорителями MAX IV, позволяют изучать фазовые переходы, отклик на внешнее воздействие и динамическую эволюцию сложных материалов, полимеров и жидкостей с использованием методов когерентного рассеяния рентгеновского излучения с временным разрешением, таких как рентгеновская фотон-корреляционная спектроскопия.
Необходимо отметить, что организаторы конференции являются участниками проекта создания в Новосибирской области (р.п. Кольцово) суперсовременного источника синхротронного излучения поколения 4+ Центр коллективного пользования «Сибирский кольцевой источник фотонов» (ЦКП «СКИФ») к концу 2023 года.
 Продолжил пленарную сессию директор Института металлургии Уральского отделения РАН в Екатеринбурге, профессор Уральского федерального университета, член правления Российского химического общества имени Д.И. Менделеева, академик РАН Андрей Андреевич Ремпель. Его лекция “Структурная диагностика с использованием рентгеновского и синхротронного излучения” была посвящена исследованиям порошков наночастиц оксидов, карбидов и сульфидов металлов, а также стеклокерамики, с включениями наночастиц люминесцентных сульфидов. В докладе были рассмотрены дифракционные методы и методы малоуглового рассеяния рентгеновских лучей для определения структуры и морфологии наноматериалов с использованием как обычных рентгеновских источников, так и синхротронного излучения.
Продолжил пленарную сессию директор Института металлургии Уральского отделения РАН в Екатеринбурге, профессор Уральского федерального университета, член правления Российского химического общества имени Д.И. Менделеева, академик РАН Андрей Андреевич Ремпель. Его лекция “Структурная диагностика с использованием рентгеновского и синхротронного излучения” была посвящена исследованиям порошков наночастиц оксидов, карбидов и сульфидов металлов, а также стеклокерамики, с включениями наночастиц люминесцентных сульфидов. В докладе были рассмотрены дифракционные методы и методы малоуглового рассеяния рентгеновских лучей для определения структуры и морфологии наноматериалов с использованием как обычных рентгеновских источников, так и синхротронного излучения.
В начале своего выступления Андрей Андреевич отметил некоторую провокационность названия своей лекции, так как в нем рентгеновское и синхротронное излучение воспринимается как разные методы. Он отметил, что в его понимании рентгеновское излучение используется в основном для идентификации атомных структур материалов, размеров наночастиц, микроструктурных характеристик, в то время как синхротронное излучение открывает значительно более широкий спектр возможностей при общности достигаемых целей. Андрей Андреевич в своей лекции также подчеркнул, что задачи, решаемые с привлечением методов рассеяния рентгеновского излучения, нужно сначала опробовать в лабораторных условиях, используя обычные рентгеновские трубки, а затем выходить на синхротроны, которые обеспечивают значительно более высокое пространственное и временное разрешение.
Профессор Ремпель представил на обзорном слайде перечень методов исследования структуры материалов, отметив, что все они имеют разное пространственное разрешение. Это методы оптической спектроскопии, зондовой микроскопии, электронной спектроскопии. Не в обиду специалистам спектрального анализа, Андрей Андреевич заметил, что электронная спектроскопия дает возможность увидеть только часть спектра, в то время как рентгеновское и синхротронное излучение позволяют увидеть материал целиком, а при необходимости сфокусироваться на определенной области материала. Нельзя забывать про динамическое рассеяние света – развитие этого метода сейчас дошло до очень высокого уровня, он позволяет наблюдать частицы размером от одного нанометра и больше, участвующие в броуновском движении в коллоидном растворе. Профессор Ремпель сконцентрировал внимание аудитории на методе малоуглового рассеяния с использованием рентгеновских лучей или нейтронных пучков, направленном на исследование именно наноструктуры материалов и имеющем высокое пространственное разрешение (от одного ангстрема до 100 нанометров). С помощью МУР исследуется широкий круг объектов, таких как наночастицы мицелл, коллоидные растворы, полимеры, вирусы, изучается распад твердых растворов, образование наночастиц в сплавах, сталях. Важно отметить, что этот метод широко используется биологами в вирусологии в целях получения безопасных вакцин. Андрей Андреевич отметил, что, хотя развитие и использование структурных методов исследования направлено на определение размеров наночастиц и распределения их по размерам, это не является самоцелью, оно преследует и более глубокие цели, такие как исследование нанаразмерных эффектов, изучение фотокаталитических и каталитических материалов, механизмов протекания каталитических реакций. Содержание лекции профессор Ремпель разделил на три части:
- широкоугловое рассеяние рентгеновских лучей (структурный и фазовый анализ, определение размеров наночастиц, величин деформации кристаллической решетки);
- малоугловое рассеяние рентгеновских лучей (атомный контраст или контраст электронной плотности, размер и форма наночастиц);
- результаты измерений на наночастицах сульфидов, карбидов и оксидов.
Подробно представив предложенные методы и продемонстрировав примеры их использования, профессор Ремпель отметил в выводах, что для точного измерения размера и формы наночастиц, их распределения по размерам необходимо использовать совокупность различных методов, включающих малоугловое рассеяние рентгеновских лучей и тепловых нейтронов. При этом результаты необходимо подтверждать электронной микроскопией, а также дифракцией рентгеновских лучей и тепловых нейтронов.
Лекцию “Рентгеноструктурный анализ монокристаллов: что химикам дает синхротрон?” представил д.х.н., профессор Виктор Николаевич Хрусталев – заведующий кафедрой неорганический химии Российского университета дружбы народов (РУДН). С 2017 года он является Директором Объединенного института химических исследований РУДН. Виктор Николаевич начал лекцию с утверждения, что использование синхротронного излучения позволяет принципиальным образом снизить требования, предъявляемые к качеству кристаллов, например, могут успешно расшифровываться достаточно мелкие (размером до 10 мкм) кристаллы с частичной разупорядоченностью. При этом существенно сокращается среднее время эксперимента, что позволяет реализовать режим высокопроизводительного скри-нинга. Синхротронное излучение практически используется во всех областях науки, в том числе и в гуманитарных науках для изучения интересных научных артефактов. Поэтому, по мнению Виктора Николаевича, и тема конференции, и срок ее проведения, назрели как никогда ранее.
Профессор Хрусталев констатировал, что, к сожалению, Россия в области использования синхротронного излучения значительно отстает от мировых ведущих стран. На сегодняшний день у нас работают только два синхротронных центра – в НИЦ "Курчатовский институт" (Москва) и в Институте ядерной физики им. Г.И. Будкера СО РАН (Новосибирск). Самые известные источники синхротронного излучения находятся в Европе, причем синхротронные центры, расположенные в основном в пригородах, имеют не только научную значимость, но и архитектурную ценность. Ведущие мировые синхротронные источники более современны и совершенны по сравнению с КИСИ и СЦСТИ, но при этом использование ускорителя в Москве и синхротронных станций на этом ускорителе все равно несопоставимо с возможностями лабораторных приборов. Поэтому создание синхротронных центров уже получило определенное развитие и привлекло внимание руководства страны. В.В. Путин в 2018 году посетил НИЦ "Курчатовский институт", потом собрал совещание с членами Академии наук, со всеми заинтересованными лицами, и на этом совещании дал указание разработать программу по развитию синхротронно-нейтронных исследований, включая создание трех новых источников синхротронного излучения: в Протвино, Новосибирске (СКИФ) и Владивостоке. На данном этапе наибольшее развитие получил проект СКИФ.
Станции, специализирующиеся в области проведения рентгеноструктурного анализа монокристаллов низкомолекулярных соединений (химической кристаллографии), являются одними из наиболее востребованных и результативных в мировых центрах СИ. В России подобная станция в составе Курчатовского источника синхротронного излучения (КИСИ, НИЦ «Курчатовский институт», Москва) появилась в 2014 году. На этой станции проводятся систематические рентгеноструктурные исследования самых разных классов кристаллических материалов, включая полиядерные каркасные металлорганосилоксаноляты, гибридные органические перовскиты, оптически чистые аналоги природных соединений, координационные соединения металлов, проявляющие каталитические, магнитные, электро- и фотолюминесцентные свойства, и т.п. В своей лекции профессор Хрусталев представил общую последовательность шагов по съемке, расшифровке и уточнению структур функциональных материалов, приводя примеры геометрически необычных и «красивых» молекулярных структур. На нескольких примерах Виктор Николаевич продемонстрировал очевидные преимущества использования источника синхротронного излучения по сравнению с современными лабораторными дифрактометрами.
 Лекция д.ф.-м.н., профессора Владимира Васильевича Чернышева “Порошковый рентгеноструктурный анализ новых материалов” была посвящена рентгеновской порошковой дифракции – методу, который уже много десятилетий считается незаменимым в области исследования новых твердофазных материалов, получаемых в различных университетских и промышленных лабораториях. Владимир Васильевич является заведующим лабораторией физико-химического анализа кафедры общей и неорганической химии Московского государственного университета имени М.В. Ломоносова. Он декларировал, что, используя легкодоступный арсенал экспериментального рентгеновского оборудования, порошковая дифракция позволяет довольно быстро проводить многие актуальные исследования, такие как классический фазовый анализ, анализ тонких и очень тонких пленок, многослойных покрытий, анализ наноразмерных порошков и материалов, анализ распределения размеров наночастиц с использованием малоуглового рассеяния рентгеновских лучей, определение фазовых переходов при изменении параметров кристаллической структуры образца, определение и уточнение параметров элементарной ячейки, определение неизвестных кристаллических структур.
Лекция д.ф.-м.н., профессора Владимира Васильевича Чернышева “Порошковый рентгеноструктурный анализ новых материалов” была посвящена рентгеновской порошковой дифракции – методу, который уже много десятилетий считается незаменимым в области исследования новых твердофазных материалов, получаемых в различных университетских и промышленных лабораториях. Владимир Васильевич является заведующим лабораторией физико-химического анализа кафедры общей и неорганической химии Московского государственного университета имени М.В. Ломоносова. Он декларировал, что, используя легкодоступный арсенал экспериментального рентгеновского оборудования, порошковая дифракция позволяет довольно быстро проводить многие актуальные исследования, такие как классический фазовый анализ, анализ тонких и очень тонких пленок, многослойных покрытий, анализ наноразмерных порошков и материалов, анализ распределения размеров наночастиц с использованием малоуглового рассеяния рентгеновских лучей, определение фазовых переходов при изменении параметров кристаллической структуры образца, определение и уточнение параметров элементарной ячейки, определение неизвестных кристаллических структур.
В своей лекции профессор Чернышев основное внимание уделил проблемам, возникающим при работе с новыми порошковыми материалами в процессе определения кристаллических структур непосредственно из порошковых дифракционных данных. Методы порошкового рентгеноструктурного анализа (пРСА) активно развиваются с начала 90-х годов прошлого века и к настоящему времени зарекомендовали себя надежным «поставщиком» новых кристаллических структур, что подтверждается десятками тысяч структур в Кембриджской кристаллографической базе и таким же количеством статей в международных научных журналах. Однако создание и развитие новых методов пРСА не избавляет от необходимости учета «старых» проблем, с которыми постоянно сталкиваются разработчики новых функциональных материалов. К числу таких проблем относятся, в частности, неоднофазность образца, слабая кристалличность новой фазы, нестабильность образца на открытом воздухе либо под воздействием других внешних факторов (например, температуры или рентгеновского излучения), чрезвычайно малые количества образца, несоответствие элементного состава образца, почти всегда содержащего и аморфную фазу, фактическому составу изучаемой кристаллической фазы.
Наряду с кратким обзором современных возможностей методов пРСА профессор Чернышев наметил пути преодоления вышеперечисленных проблем в процессе работы с новыми материалами и установления кристаллической структуры новой «нужной» фазы. Хотя современные лабораторные порошковые дифрактометры способны предоставить надежный экспериментальный материал для решения подобных задач, Владимир Васильевич продемонстрировал критически важное значение синхротронной дифракции высокого разрешения в структурной характеризации новых материалов. В заключение он привел практические примеры работы с различными материалами, такими как металл-органические каркасы (MOF) и нанопористые углеродные материалы на их основе, органические каркасы, образованные за счет водородных (HOF) или ковалентных (COF) связей, комплексы тетрапиррольных макроциклов с редкоземель-ными элементами, металлсодержащие цеолиты, бета-замещенные металлопорфирины, тройные интерметаллиды с редкоземельными элементами, оптически активные производные терефталевой кислоты, хиральные фосфиты, фармацевтические субстанции.
 Заключительный день работы конференции не ослабил внимание участников к пленарной сессии. С лекцией “Исследование магнитных свойств функциональных материалов” выступил директор Института физики им. Л.В. Киренского Сибирского отделения РАН из Красноярска, д.ф.-м.н. Дмитрий Александрович Балаев, известный в мире специалист в области физики магнитных явлений. Дмитрий Александрович отметил тот факт, что исследование магнитных свойств является неотъемлемой частью характеризации получаемых функциональных материалов, содержащих магнитные ионы, магнитоупорядоченные фазы, магнитные наночастицы. На примере ряда объектов он проиллюстрировал возможности применения «магнитной характеризации» для получения информации о магнитном состоянии материала, магнитной структуре материала, фазовом составе материала, наличии магнитных примесей. Особое внимание в докладе было уделено магнитным наночастицам (оксиду никеля, модификациям оксида железа), их магнитной структуре, влиянию магнитных межчастичных взаимодействий, а также поверхностным и размерным эффектам, проявляющимся в магнитных свойствах. Дмитрий Александрович представил обзор подходов к описанию магнитных свойств наночастиц ферро-, ферри- и антиферромагнитных материалов. При этом он заметил, что, возможно, его лекция в большей степени будет интересна участникам, не специализирующимся в области физики магнитных явлений.
Заключительный день работы конференции не ослабил внимание участников к пленарной сессии. С лекцией “Исследование магнитных свойств функциональных материалов” выступил директор Института физики им. Л.В. Киренского Сибирского отделения РАН из Красноярска, д.ф.-м.н. Дмитрий Александрович Балаев, известный в мире специалист в области физики магнитных явлений. Дмитрий Александрович отметил тот факт, что исследование магнитных свойств является неотъемлемой частью характеризации получаемых функциональных материалов, содержащих магнитные ионы, магнитоупорядоченные фазы, магнитные наночастицы. На примере ряда объектов он проиллюстрировал возможности применения «магнитной характеризации» для получения информации о магнитном состоянии материала, магнитной структуре материала, фазовом составе материала, наличии магнитных примесей. Особое внимание в докладе было уделено магнитным наночастицам (оксиду никеля, модификациям оксида железа), их магнитной структуре, влиянию магнитных межчастичных взаимодействий, а также поверхностным и размерным эффектам, проявляющимся в магнитных свойствах. Дмитрий Александрович представил обзор подходов к описанию магнитных свойств наночастиц ферро-, ферри- и антиферромагнитных материалов. При этом он заметил, что, возможно, его лекция в большей степени будет интересна участникам, не специализирующимся в области физики магнитных явлений.
В рубрике «Методы измерения намагниченности» профессор Балаев дал краткую информацию о магнитных полях в природе, затем перешел к вопросу – как и чем их можно создать, представив слайд о времени изготовления и затратах, охарактеризовал основные приборы для измерения магнитных свойств в различных диапазонах величин магнитных полей. Самым популярным и наиболее часто используемым в этих целях прибором Дмитрий Александрович считает вибрационный магнетометр, обладающий высокой точностью. Но тем не менее, самым точным он назвал СКВИД магнетометр – в нем сигнал от образца фиксируется датчиком, в котором есть джозефсоновский контакт (S-I-S). Были даны характеристики и другим магнетометрам, включая импульсные, обладающим невысокой точностью и специфичностью.
Далее Дмитрий Александрович обрисовал основные типы и характеристики магнитных материалов, а также более сложных систем на их основе. Он отметил характерные особенности, присущие ферро-ферримагнитным наночастицам при уменьшении размеров и увеличении доли поверхностных атомов. На примере оксида железа были рассмотрены вопросы перехода в однодоменное состояние, супермагнетизм, суперпарамагнитная блокировка, а также – к чему приводит уменьшение размера частиц (поверхностная анизотропия, размерные эффекты), магнитные межчастичные взаимодействия. Обсужден «отрицательный» эффект большой поверхности и указан метод, предложенный совместно с коллегами из ИК СО РАН, позволяющий преодолеть эту проблему.
В заключение Дмитрий Александрович предложил рассмотреть еще один пример, иллюстрирующий, как можно идентифицировать очень малое количество неизвестного соединения с помощью магнитных измерений. Были исследованы магнитные свойства донных отложений соленого меромиктического озера Шира в Хакасии. В них впервые проведены измерения магнитных свойств методом статической магнитометрии и γ-резонансной мёссбауэровской спектроскопии. Во всех слоях донных отложений присутствуют наноразмерные однодоменные частицы магнетита, источником которых являются магнитотактические бактерии. Содержание магнетита в донных отложениях убывало с глубиной, при этом обнаружен локальный минимум в слое, соответствующем минимальному уровню озера, наблюдавшемуся в 1910–1930 гг. Было показано, что биогенный магнетит может служить, наряду с прочими биологическими и геохимическими характеристиками, индикатором климатически обусловленных изменений уровня воды в озере Шира.
Выводы, представленные Дмитрием Александровичем Балаевым, показали, что магнитометрия является мощным методом характеризации и исследований магнитной структуры и фундаментальных параметров функциональных материалов, а системы магнитных наночастиц являются ярким примером влияния поверхности и размерных эффектов на магнитную структуру и макроскопические свойства частиц.
 С большим интересом аудитория встретила лекцию к.ф.-м.н. Александра Леонидовича Васильева из НИЦ "Курчатовский институт" и ФНИЦ "Кристаллография и фотоника" (Москва), завершавшую пленарную сессию: “Электронная микроскопия в исследовании функциональных материалов”.
С большим интересом аудитория встретила лекцию к.ф.-м.н. Александра Леонидовича Васильева из НИЦ "Курчатовский институт" и ФНИЦ "Кристаллография и фотоника" (Москва), завершавшую пленарную сессию: “Электронная микроскопия в исследовании функциональных материалов”.
Просвечивающая электронная микроскопия высокого разрешения (HRTEM) и сканирующая просвечивающая электронная микроскопия высокого разрешения (HRSTEM) вместе с энергодисперсионным рентгеновским микроанализом (EDX) и спектроскопией потерь энергии электронов (EELS) во многих случаях являются единственными инструментами для определения особенностей кристаллической структуры. Эти методы уникальны для характеризации кристаллической структуры при неоднозначном определении фазы, если размеры частиц находятся в нанометровом диапазоне, а также в условиях наличия протяженных и особенно точечных дефектов кристаллической структуры. Использование корректоров сферической аберрации (Cs) улучшает пространственное разрешение HRTEM и HRSTEM до менее 0,1 нм. Это позволяет наблюдать практически все кристаллические структуры с атомным разрешением в различных низко-индексовых зонах. Использование дифракции электронов (ЭД) и даже ЭД сходящегося пучка для определения кристаллической структуры может быть затруднительным при наличии дефектов кристалла, что снижает симметрию дифракционной картины и вносит неопределенность в определение пространственной группы неизвестной фазы. Исследование неизвестных фаз и даже точечных дефектов может быть выполнено с помощью широкоуглового кольцевого детектора темного поля (HAADF) в HRSTEM. Это единственный метод точного определения точечных дефектов, включая положение вакансий или атомных замещений и их плотность.
В своей лекции Александр Леонидович продемонстрировал несколько примеров определения фаз и исследования точечных дефектов в различных материалах. При этом он отметил, что задача определения новых фаз в поликристаллических мелкодисперсных или нано-материалах методами рентгеноcтруктурного анализа и рентгенофазового анализа часто невозможна из-за малых размеров частиц неизвестной фазы, присутствия структурных дефектов, большого набора присутствующих в образцах дифракционных максимумов, ассоциированных с различными фазами, в том числе низко симметричными, а также различной ориентации частиц. В таких случаях единственной возможностью решения задачи определения кристаллической структуры может быть использование просвечивающей/растровой электронной микроскопии (П/РЭМ) высокого разрешения (ВР) и электронной дифракции (ЭД). При размерах частиц менее 10 нм практически невозможно использование селективной диафрагмы для получения высококачественной электронограммы от выделенной области. В этом случае приходится использовать методы дифракции в сходящемся пучке, микро- и нано-дифракцию. В этих условиях происходит умень-шение точности определения параметров элементарной ячейки. Более того, присутствие структурных дефектов в частицах понижает симметрию дифракционной картины и вносит неопределенности в определение пространственной группы неизвестной фазы. Использование П/РЭМ ВР позволяет выявить структурные дефекты и значительно повысить точность определения геометрических параметров кристалла. Причем, если использование ПЭМ ВР требует моделирования изображения, то темнопольное ПРЭМ ВР с регистрацией электронов, рассеянных на большие углы, часто легко интерпретируется и не нуждается в моделировании. В сочетании с энергодисперсионным рентгеновским микроанализом (ЭРМ) высокого разрешения и/или спектроскопией потерь энергии электронов (СПЭЭ), интерпретация становится еще проще – удается установить структуру с определением атомных позиций, а также состав основной фазы и дефектов с атомным разрешением.
Продемонстрировав аргументированные примеры, Александр Леонидович сделал заключение, что использование высокоразрешающих методов ПЭМ/ПРЭМ существенно помогает в определении ряда структурных особенностей кристаллов. Но при этом он счел необходимым заметить, что применение других структурных и спектроскопических методов часто все-таки бывает необходимо для однозначной интерпретации результатов электронно-микроскопических исследований.
Ключевая сессия конференции МИССФМ-3 была представлена следующими лекциями:

|
К.ф.-м.н. Солдатов Михаил Александрович МИЦ "Интеллектуальные материалы", Южный федеральный университет, Ростов-на-Дону Комбинированное использование рентгеноабсорбционной спектроскопии, флуоресцентного анализа и ИК-спектроскопии с микро- и нанопучками для исследования биомедицинских материалов |

|
Д.х.н., профессор Михлин Юрий Леонидович Институт химии и химической технологии СО РАН, Красноярск Рентгеновская фотоэлектронная спектроскопия (РФЭС) как метод изучения поверхности и реакций сульфидов металлов |

|
Д.ф.-м.н. Авдеев Михаил Васильевич Объединенный институт ядерных исследований, Дубна, Московская область Нейтронное рассеяние для магнитных материалов |

|
К.ф.-м.н. Володин Владимир Алексеевич Институт физики полупроводников им. А.В. Ржанова СО РАН, Новосибирск Определение доли кристаллической фазы в пленках функциональных материалов и размеров нанокристаллов из анализа спектров комбинационного рассеяния света |

|
х.н. Лапина Ольга Борисовна Институт катализа им. Г.К. Борескова СО РАН, Новосибирск Современные возможности ЯМР спектроскопии в твердом теле для исследования функциональных материалов |

|
Д.х.н., профессор Зверева Ирина Алексеевна Санкт-Петербургский государственный университет, Санкт-Петербург Термический анализ для исследования твердофазных процессов и диагностики функциональных материалов |
Поскольку идея проведения форума была связана с началом активной фазы проектирования Центра коллективного пользования «Сибирский кольцевой источник фотонов» (СКИФ), одной из ключевых тем конференции МИССФМ-3 стало использование синхротронного излучения (СИ). Участниками были представлены интересные доклады, посвященные методам, которые применяются при исследовании материалов с использованием СИ. В их презентациях были затронуты вопросы, связанные со структурной диагностикой материалов различного назначения с использованием рентгеновского и синхротронного излучения, использованием рентгеноструктурного анализа монокристаллов в химии, методами исследования быстропротекающих структурных превращений с высоким временным разрешением на пучках СИ, возможностям XAFS-спектроскопии для исследования функциональных наноматериалов. В докладах были представлены концептуальные проекты будущих станций ЦКП «СКИФ» «Быстро-протекающие процессы», «Структурная диагностика» и «Диагностика в высокоэнергетическом рентгеновском диапазоне». В целом, в рамках устной сессии доклады были представлены на восьми секциях:
Секция I.
Перспективные направления диагностики состава и структуры функциональных материалов, в том числе, с использованием синхротронного излучения
Секция II.
ИК-, КР-спектроскопия и методы определения химического состава функциональных материалов на макро-, микро- и наноуровне
Секция III.
Методы изучения магнитных свойств функциональных материалов
Секция IV.
Методы определения параметров кристаллической структуры и наноструктуры
Секция V.
Методы определения дисперсности и текстурных характеристик
Секция VI.
Комплексные исследования структуры и свойств материалов
Секция VII.
Методы определения электронных характеристик вещества
Секция VII.
Термоаналитические методы
Особое внимание было привлечено к Секции молодых ученых, на которой аспиранты и студенты-магистранты представили результаты своих работ в десятиминутных устных докладах. Презентации начинающих исследователей вызвали большой интерес со стороны аудитории. Вопросы опытных экспертов, активные дискуссии поддерживали тонус молодых ученых и мотивировали их к дальнейшей плодотворной научной деятельности
Не меньший интерес вызвала и стендовая сессия. Часть работ была представлена в формате пятиминутных флэш-презентаций, позволявших отразить ключевую направленность доклада и продемонстрировать несколько слайдов. Остальные стендовые доклады были заранее размещены на сайте конференции в формате PDF презентаций. С ними участники могли ознакомиться еще до конференции. К открытию конференции был создан онлайн-форум в Google Group со ссылками на PDF презентации, на котором была возможность обсудить доклады с автором в интерактивном режиме. Окончательное обсуждение состоялось на двухчасовом заседании стендовой сессии в конце первого дня работы конференции.
Нельзя не отметить, что пристальное внимание аудитории привлекли интересные презентационные доклады, которые были насыщены информативностью и глубиной погружения в представляемый материал. Они оказались весьма полезными для участников и вызвали большое количество вопросов, предложений к сотрудничеству и использованию полученных данных. Председатели заседаний были вынуждены призывать заинтересованных участников продолжать дебаты и переговоры о сотрудничестве с авторами докладов в индивидуальном режиме.
 Руководитель отдела «Природные ресурсы и дифрактометрия» ООО «Техноинфо» Олег Евгеньевич Корнейчик анонсировал возможности лабораторного оборудования компании и его применение в едином комплексе с синхротроном в свете последних достижений в области методов мало- и широкоуглового рентгеновского рассеяния, а также монокристальной дифракции Rigaku Oxford diffraction. Прозвучали предложения рассмотреть возможности интегрирования универсальных лабораторных систем компании ТехноИнфо в проект СКИФ, что вызвало незамедлительный ответный интерес со стороны главного эксперта Проектного офиса ЦКП «СКИФ», заведующего лабораторией перспективных синхротронных методов исследования ИК СО РАН, председателя Программного комитета конференции МИССФМ-3, д.ф.-м.н. Яна Витаутасовича Зубавичуса.
Руководитель отдела «Природные ресурсы и дифрактометрия» ООО «Техноинфо» Олег Евгеньевич Корнейчик анонсировал возможности лабораторного оборудования компании и его применение в едином комплексе с синхротроном в свете последних достижений в области методов мало- и широкоуглового рентгеновского рассеяния, а также монокристальной дифракции Rigaku Oxford diffraction. Прозвучали предложения рассмотреть возможности интегрирования универсальных лабораторных систем компании ТехноИнфо в проект СКИФ, что вызвало незамедлительный ответный интерес со стороны главного эксперта Проектного офиса ЦКП «СКИФ», заведующего лабораторией перспективных синхротронных методов исследования ИК СО РАН, председателя Программного комитета конференции МИССФМ-3, д.ф.-м.н. Яна Витаутасовича Зубавичуса.
 В свою очередь, Ян Витаутасович Зубавичус от имени редколлегии журнала Crystals, являющегося медиа-партнером конференции МИССФМ-3, выступил с презентацией этого журнала, а также издательства Multidisciplinary Digital Publishing Institute (MDPI), Швейцария, в котором Crystals издается. Ян Витаутасович является членом редколлегии журнала Crystals по секции Organic Crystalline Materials, и в данный момент – приглашенным редактором специального выпуска «Текущие вызовы кристаллографии, фокусированные на структурах промежуточной сложности». В первую очередь Ян Витаутасович рассказал об издательстве MDPI, которое является одним из ведущих издательств в мире, специализирующихся в области электронной публикации научно-технической литературы. Базируется MDPI в Базеле, Швейцария, имея филиалы во многих странах. Издательство очень активно развивается, придерживаясь концепции открытого доступа (open access). Это предполагает бесплатный, быстрый, полнотекстовый доступ в режиме реального времени к научным и учебным материалам, реализуемый для любого пользователя в глобальной информационной сети без каких-либо ограничений как по инструментам доступа, так и по дальнейшему использованию опубликованных материалов (обязательна точная ссылка на цитируемый источник, но дополнительного разрешения издательства не требуется). Режим открытого доступа применяется ко всем формам опубликованных результатов исследований, в том числе к статьям рецензируемых и нерецензируемых научных журналов, материалам конференций, текстам диссертаций и квалификационным работам, главам книг и монографиям. Далее Ян Витаутасович перешел к рассказу о медиа-партнере конференции, журналу Crystals, одному из ведущих источников по исследованиям кристаллических материалов. Концепция Open Access позволяет журналу сократить время опубликования статей. С момента подачи рукописи до принятия решения оно составляет 13 дней при жестком режиме рецензирования. Ян Витаутасович привел некоторые статистические данные по публикациям в журнале, отразил тематику публикаций, которые близки к научным направлениям конференции МИССФМ-3. Поэтому участники были приглашены представить статьи в журнал Crystals, а также в специальный выпуск, редактируемый Яном Витаутасовичем. Как медиа-спонсор, журнал Crystals предоставил участникам конференции МИССФМ-3 25%-ю скидку на публикацию статей.
В свою очередь, Ян Витаутасович Зубавичус от имени редколлегии журнала Crystals, являющегося медиа-партнером конференции МИССФМ-3, выступил с презентацией этого журнала, а также издательства Multidisciplinary Digital Publishing Institute (MDPI), Швейцария, в котором Crystals издается. Ян Витаутасович является членом редколлегии журнала Crystals по секции Organic Crystalline Materials, и в данный момент – приглашенным редактором специального выпуска «Текущие вызовы кристаллографии, фокусированные на структурах промежуточной сложности». В первую очередь Ян Витаутасович рассказал об издательстве MDPI, которое является одним из ведущих издательств в мире, специализирующихся в области электронной публикации научно-технической литературы. Базируется MDPI в Базеле, Швейцария, имея филиалы во многих странах. Издательство очень активно развивается, придерживаясь концепции открытого доступа (open access). Это предполагает бесплатный, быстрый, полнотекстовый доступ в режиме реального времени к научным и учебным материалам, реализуемый для любого пользователя в глобальной информационной сети без каких-либо ограничений как по инструментам доступа, так и по дальнейшему использованию опубликованных материалов (обязательна точная ссылка на цитируемый источник, но дополнительного разрешения издательства не требуется). Режим открытого доступа применяется ко всем формам опубликованных результатов исследований, в том числе к статьям рецензируемых и нерецензируемых научных журналов, материалам конференций, текстам диссертаций и квалификационным работам, главам книг и монографиям. Далее Ян Витаутасович перешел к рассказу о медиа-партнере конференции, журналу Crystals, одному из ведущих источников по исследованиям кристаллических материалов. Концепция Open Access позволяет журналу сократить время опубликования статей. С момента подачи рукописи до принятия решения оно составляет 13 дней при жестком режиме рецензирования. Ян Витаутасович привел некоторые статистические данные по публикациям в журнале, отразил тематику публикаций, которые близки к научным направлениям конференции МИССФМ-3. Поэтому участники были приглашены представить статьи в журнал Crystals, а также в специальный выпуск, редактируемый Яном Витаутасовичем. Как медиа-спонсор, журнал Crystals предоставил участникам конференции МИССФМ-3 25%-ю скидку на публикацию статей.
 Мастер-класс “Использование онлайн приложения автоматического распознавания наночастиц в изображениях сканирующей зондовой микроскопии с применением подходов искусственного интеллекта и машинного обучения”. Огромный интерес вызвал мастер-класс, который провела старший научный сотрудник Института катализа им. Г.К. Борескова СО РАН, доцент кафедры химии твердого тела Факультета естественных наук Новосибирского государственного университета, к.х.н. Анна Владимировна Нартова. В нем шла речь о разработанной программе автоматического распознавания наночастиц в изображениях атомно-силового микроскопа с использованием подходов искусственного интеллекта и машинного обучения. Программа оформлена в виде свободного веб-интерфейса: можно зайти на сайт, загрузить свою картинку и получить статистику по наночастицам (количество, средний размер, степень заполнения поверхности и пр.). Анна Владимировна провела краткий мастер-класс по практическому использованию данной программы.
Мастер-класс “Использование онлайн приложения автоматического распознавания наночастиц в изображениях сканирующей зондовой микроскопии с применением подходов искусственного интеллекта и машинного обучения”. Огромный интерес вызвал мастер-класс, который провела старший научный сотрудник Института катализа им. Г.К. Борескова СО РАН, доцент кафедры химии твердого тела Факультета естественных наук Новосибирского государственного университета, к.х.н. Анна Владимировна Нартова. В нем шла речь о разработанной программе автоматического распознавания наночастиц в изображениях атомно-силового микроскопа с использованием подходов искусственного интеллекта и машинного обучения. Программа оформлена в виде свободного веб-интерфейса: можно зайти на сайт, загрузить свою картинку и получить статистику по наночастицам (количество, средний размер, степень заполнения поверхности и пр.). Анна Владимировна провела краткий мастер-класс по практическому использованию данной программы.
Проведение мероприятия в формате онлайн стало первым опытом в широком спектре организуемых Институтом катализа конференций, традиционно проходящих в очном режиме. Приятно осознавать, что организаторам удалось создать атмосферу, которая позволила большому количеству участников, находящихся в разных частях света (от Германии, Швеции, Калининграда до Читы и Владивостока) стать частью великолепного форума, окунуться в волну новой информации, дебатов, споров, получить удовольствие от общения друг с другом, и даже насладиться запоминающимся фуршетом с бокалом вина, музыкой, песнями под гитару, воспоминаниями.

На закрытии конференции участники отметили высокий уровень пленарной и ключевой сессий, четкую организацию всех научных сессий с представлением интересных устных, стендовых, презентационных докладов, согласованную и слаженную работу научно-технических модераторов, уникальную атмосферу, которая царила во время проведения конференции.
Диплом за лучший стендовый доклад был единогласно присужден доценту Кемеровского государственного университета, к.х.н. Наталье Владимировне Ивановой: «Применение вольтамперометрических методов в анализе наноструктури¬ро-ванных биметаллических систем».
Диплома за лучший устный доклад среди молодых ученых был удостоен старший научный сотрудник Института геологии и геохимии УрО РАН (Екатеринбург), к.г.-м.н. Дмитрий Александрович Замятин: «Исследование неоднородного строения кристаллов совместным анализом электронных и оптических изображений».
Тезисы всех презентаций доступны по ссылке: http://catalysis.ru/resources/institute/Publishing/Report/2020/abstracts-missfm-2020.pdf
Запись всех презентаций доступна по ссылке:http://conf.nsc.ru/missfm-3/ru/missfm-3_Vid
Отобранные Программным комитетом статьи будут опубликованы в Журнале Структурной химии.
Материал подготовили:
А.А. Ведягин, Я.В. Зубавичус, С.В. Цыбуля, Т.В. Замулина
(Институт катализа СО РАН, Новосибирск)
Chemists shine light on new way to think about reductive elimination
Computational study confirms alternative mechanism for C-C bond forming reaction
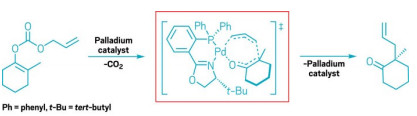
The Pd-catalyzed allylic alkylation goes through a transition state involving 7 atoms.
Reductive elimination reactions, in which a transition metal catalyzes new bond formation between two molecules, are one of organic chemists’ most powerful tools. For example, one flavor of these reactions, palladium-catalyzed allylic alkylations can make new C–C bonds while tolerating many functional groups. In addition, these mild reactions are one of the few ways chemists can generate compounds containing hindered quater-nary carbons.
Through a detailed computational study, Brian Stoltz and coworkers at the California Institute of Technology have figured out that some Pd-catalyzed reductive eliminations go through a different mechanism than previously thought, providing a new understanding of how reductive eliminations work (J. Am. Chem. Soc., 2020, DOI: 10.1021/jacs.0c09575).
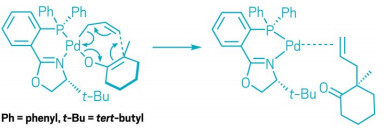
Based on their computational work, chemists built a familiar arrow pushing diagram
to explain what happens in the transition state of the Pd-catalyzed allylic alkylation.
Traditionally, chemists thought the mechanism involved transition states featuring bond breaking and forming between three key atoms. Stoltz and coworkers studied a specific Pd-catalyzed reductive elimination reaction in which the desired C-C bond forms within the same molecule (shown). The chemists determined that the transition state for this reaction involves bond forming and breaking between seven atoms. Describing this seven-centered reductive elimination process allows chemists to expand the way they think about this fundamental reaction, Stoltz says. This mechanism is not the way chemists learn how reductive eliminations work in graduate school, he says, but it suggests that there are other ways that reductive elimination can happen. “It could change the way people think about what’s possible.”
For this computational study, the researchers focused solely on the Pd-centered transition state. Generally, this transition state is difficult for chemists to study experimentally because it’s not the rate-limiting step of the reaction, so it exists briefly. Chemists have suspected that this transi-tion state involves 7 key atoms, and is organized in a ring, like those found in pericyclic reactions. Not only did Stoltz and coworkers confirm this suspicion, but also they showed that the Pd’s d-orbitals allow the transition state to be aromatic, which means it’s stabilized through ex-tended conjugation of orbitals across the seven atoms. This stabilization allows the transition-state energy to be low enough for the reaction to proceed. In addition, they found that because of the geometry of Pd’s d orbitals this aromaticity doesn’t fit the traditional structure, instead it is shaped like a Möbius strip, which is a connected loop with one surface and a half turn.
The researchers also used a type of chemical bonding theory to explain the mechanism of the reaction in terms of arrow pushing. This drawing method in which arrows depict where electrons move in a chemical reac-tion is the way that many organic chemists are taught to understand these transformations.
Through detailed analysis, the researchers showed the inner workings of an important organometallic reaction, says Dean Tantillo, a computational organic chemist at the University of California, Davis. They used hard-core quantum chemistry to build a model using familiar, intuitive concepts, he says.
This study “allows us to think about some of the things we can improve on,” in the current reaction, and gives a high-level theoretical backing for the feasibility of new kinds of reaction, Stoltz says.
TiO₂ nanocrystals exhibit unusual slow-motion blinking
Made via template-based synthesis, the defective crystals actively mediate photocatalysis
Like microscopic fireflies, semiconductor nanocrystals, also known as quantum dots, light up intermittently. For more than 20 years, researchers have worked to understand and control these random fluctuations in light emission because the “blinking” limits the stability of quantum dot–based devices like solar cells and light-emitting diodes. The observation of what may be a new type of blinking behavior suggests that researchers have more work to do (Angew. Chem., Int. Ed. 2020, DOI: 10.1002/anie.202005143). A team led by Tao Zhang, Tewodros Asefa, and Alexei M. Tyryshkin of Rutgers University and Eliška Mikmeková of the Czech Academy of Sciences reports that treating a polymer-derived porous carbon template with a titanium dioxide precursor yields TiO2 nanocrystals—limited to 10 nm in diameter and riddled with oxygen vacancies—that blink in an unprecedented way. In contrast to years of studies reporting rapid light–induced blinking with flashes often lasting just a fraction of a second—sometimes longer—the new work describes electron beam–induced flashes lasting roughly 15 s. In addition, when the crystals are irradiated with light, the resulting charge separation is unusu-ally stable, making the particles active photocatalysts, as shown by tests in which the team used them to reduce carbon dioxide with water.
Road to chiral alkylamines paved with iridium
Cationic catalyst adds amines to internal alkenes
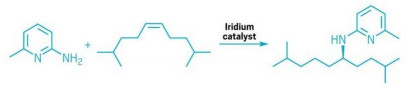
Alkylamines are one of the most useful compounds in a chemist’s bag of tricks. Pharmaceuticals, agrochemicals, and polymers often contain these functional groups. Ideally, chemists would like to make alkylamines from widely available feedstocks, but the existing methods for doing so don’t excel at reacting with internal alkenes. John Hartwig and coworkers at the University of California, Berkeley, have now figured out how. The group used a positively charged iridium catalyst and an amine to add an NH bond to an internal alkene. This generates chiral alkylamines with high regioselectivity and moderate to high yields (Nature, 2020, DOI: 10.1038/s41586-020-2919-z).
Researchers have tried to make similar compounds in the past but have not been able to get good yields of the desired chiral products. The key to Hartwig’s synthesis was choosing bidentate aminopyridine derivatives both as ligands and reactants, which helps control the chirality of the product while preventing unwanted side reactions. During the reaction, two aminopyridine molecules bind to the Ir center. One is displaced by the incoming alkene while the other remains on the catalyst. The researchers think this configuration locks the geometry so the alkene can only add from one side of the catalyst molecule. The amine group on the aminopyridine helps to block unwanted side reactions, leading to chiral alkylamines with regioselectivity near 100% in some cases. The pyridine moiety can then be removed to give the final amine.
The cationic Ir catalyst also speeds up the reductive elimination step to release the alkylamine, which “leads to addition faster than isomerization,” Hartwig says. The team hopes that this new synthesis will lead to adding O–H and C–H groups in the same way.
The ultimate goal is to use ammonia as the amine source, but the researchers started out with the aminopyridine as a stand-in. “This approach is promising because it not only tunes the catalyst but includes an ammo-nia surrogate,” says Vy Dong, an organic chemist at the University of California, Irvine. “It highlights the power of designing reagents and using mechanistic insights to achieve otherwise impossible reactivity.”
One-pot method turns polyethylene into valuable detergent precursor
Pt-catalyzed upcycling process doesn’t use solvents or relatively high temperatures
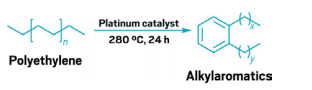
Though polyethylene (PE) is recyclable, the high-temperature, energy-intensive process yields only low-value products. As a result, industries don’t earn enough money to make recycling PE worth their effort. Now Susannah Scott and coworkers from the University of California, Santa Barbara; the University of Illinois; and Cornell University have developed a catalytic process to upcycle PE into something of higher value: long-chain alkylaromatics (Science 2020, DOI: 10.1126/science.abc5441). Scientists can sulfonate these compounds to make detergents and surfactants. The market for one of the alkylaromatics, linear alkylbenzenes, is about $9 billion per year. To upcycle PE, the researchers seal either high- or low-density PE and an alumina-supported platinum catalyst in a reaction vessel. They then heat the materials to 280 °C for 24 h. The polymer melts and then reacts with the catalyst, releasing H2. This hydrogenolysizes the PE, and these smaller bits of polymer then aromatize to give the higher-value products in 80% yield. This method “couples a thermodynamically unfavorable reaction, the aromatization, with a thermodynamically favorable reaction, the hydrogenolysis,” Scott says, a key step to getting the temperature below 300 °C.
Chemical & Engineering News
|
24-27
февраля 2021 г. Международная научная конференция «Техника и технология нефтехимического и нефтегазового производства: химическая технология – 2021» (Oil and Gas Engineering: Chemical Technology – 2021) Омск, Россия |
http://oge.omgtu.ru/ |
|
Spring
or Autumn 2021 6th European Symposium on Photopolymer Science İstanbul, Turkey |
http://www.esps2020.org/ |
|
Spring
2021 First International Symposium on High-Throughput Catalysts Design (HTCD 2020) Villeneuve D'Ascq, France |
https://htcd2020.sciencesconf.org/ |
|
March 21-24, 2021
11th International Symposium on Catalysis in Multiphase Reactors & 10th International Symposium on Multifunctional Reactors (CAMURE 11 & ISMR 10) Milan, Italy |
https://www.aidic.it/camure11-ismr10/ |
|
April
26-30, 2021 IV Scientific-Technological Symposium “Catalytic Hydroprocessing in Oil Refining” (STS HydroCat-2021) Thessaloniki, Greece |
http://conf.nsc.ru/STS_4/en |
|
May 16-19, 2021
6th International
School-Conference on Catalysis for Young Scientists “Catalyst Design: From Molecular to Industrial Level” Novosibirsk, Russia |
http://conf.nsc.ru/catdesign2021/en/ |
|
May
16-20, 2021 48th World Polymer Congress (MACRO2020) Jeju Island, Korea |
http://www.macro2020.org |
|
May
17-19, 2021 New Trends in Polymer Science (Polymers 2020) Turin, Italy |
https://polymers2020.sciforum.net/ |
|
May
19-22, 2021 2nd International Conference on Reaction Kinetics, Mechanisms and Catalysis (RKMC 2021) Budapest, Hungary |
https://rkmc.akcongress.com/ |
|
May-June,
2021 10th International Symposium “Molecular Order and Mobility in Polymer Systems” (MOMPS-X) Saint Petersburg, Russia |
http://momps2020.macro.ru/ |
|
June,
2021 11th European Conference on Solar Chemistry and Photocatalysis: Environmental Applications (SPEA11) Turin, Italy |
http://www.spea11.unito.it/home |
|
June
14-18, 2021 6th International Conference “Catalysis for Renewable Sources: Fuel, Energy, Chemicals” (CRS-6) Lisbon, Portugal |
http://conf.nsc.ru/CRS6/en |
|
21-25
июня 2021 г. XI Научно-практическая конференция «Сверхкритические флюиды: фундаментальные основы, технологии, инновации» Новосибирск, Россия |
http://scftec.isc-ras.ru/ |
|
28
июня – 2 июля 2021 г. VI Мемориальный семинар «Гомогенные и закрепленные металлокомплексные катализаторы для процессов полимеризации и нефтехимии» Листвянка, Иркутская область, Россия |
http://conf.ict.nsc.ru/ermak-VI/ru |
|
June
– July, 2021 International BioEPR School-conference Novosibirsk, Russia |
http://www.bioepr2020.ru |
|
July
4-7, 2021 8th Conference of the Federation of European Zeolite Associations “Nanoporous Materials” (FEZA 2021) Brighton, UK |
http://www.fezaconference.org |
|
July
18-22, 2021 84th Prague Meeting on Macromolecules – Frontiers of Polymer Colloids Prague, Czech Republic |
https://www.imc.cas.cz/sympo/84pmm/ |
|
July
19-23, 2021 2nd International Conference on NonCovalent Interactions (ICNI2021) Strasbourg, France |
http://icni2021.unistra.fr/ |
|
July
25-30, 2021 12th Triennial Congress of the World Association of Theoretical and Computational Chemists (WATOC 2020) Vancouver, Canada |
http://watoc2020.ca |
|
August
13-20, 2021 48th World Chemistry Congress Montreal, Canada |
https://www.cheminst.ca/ conference/ccce2021/ |
|
6-10
сентября 2021 г. XIV Конференция «Металлургия цветных, редких и благородных металлов» в рамках XII Международного конгресса и выставки «Цветные металлы и минералы» Красноярск, Россия |
https://nfmsib.ru/ |
|
September
12-15, 2021 4th European Conference on Metal Organic Frameworks and Porous Polymers (EuroMOF 2021) Krakow, Poland |
https://dechema.de/en/EuroMOF2021.html |
|
September
12-17, 2021 XXIV International Conference on Chemical Reactors CHEMREACTOR-24 Milan, Italy |
http://conf.nsc.ru/CR-24/en/ |
|
September
19-23, 2021 13th European Congress of Chemical Engineering and 6th European Congress of Applied Biotechnology (ECCE 13 & ECAB 6) Berlin, Germany |
http://ecce-ecab2021.eu/ |
|
20-25
сентября 2021 г. IV Российский конгресс по катализу «РОСКАТАЛИЗ» Казань, Россия |
http://conf.nsc.ru/RusCat-2021/ru |
|
13-16
октября 2021 г. Международная научно-техническая конференция «КАТАЛИЗ: переработка углеводородного сырья и экология» Ташкент, Узбекистан |
http://conf.nsc.ru/tashkent-2020/ru |
|
17-20
October, 2021 4th International Symposium on Multiscale Multiphase Process Engineering (MMPE) Berlin, Germany |
https://dechema.de/en/mmpe2020.html |
|
December
16-21, 2021 International Chemical Congress of Pacific Basin Societies (PAC CHEM) Honolulu, Hawaii, USA |
http://www.chemistry.or.jp/en/ events/pacifichem.html |
|
2021
г. ХI Всероссийская научная конференция и школа «Аналитика Сибири и Дальнего Востока», посвященная 100-летию со дня рождения И.Г. Юделевича Новосибирск, Россия |
http://conf.nsc.ru/asfe-11 |
|
|
|
|
July
18-22, 2022 26th IUPAC International Conference on Chemistry Education (ICCE 2020) Cape Town, South Africa |
https://iupac.org/event/26th-iupac-international-conference-on-chemistry-education/ |
|
August
28 – September 2, 2022 44th International Conference on Coordination Chemistry Rimini, Italy |
https://www.iccc2020.com/ |
|
September,
2022 7th International Conference on Metal-Organic Frameworks and Open Framework Compounds Dresden, Germany |
https://dechema.de/en/MOF2020.html |
|
September
– October, 2022 9th IUPAC International Conference on Green Chemistry (ICGC-9) Athens, Greece |
http://www.greeniupac2020.org |
|
2022-2023 European Congress on Catalysis (EuropaCat 2021) Prague, Czech Republic |
www.europacat2021.eu |
Памяти Баира Сыдыповича БАЛЬЖИНИМАЕВА
(1950 – 2020 гг.)

19 октября 2020 года скоропостижно ушёл из жизни крупный специалист в области гетерогенного катализа д.х.н. Баир Сыдыпович Бальжинимаев, внесший неоценимый вклад в развитие Института катализа СО РАН.
Б.С. Бальжинимаев родился в 1950 году. В 1973 году окончил факультет естественных наук Новосибирского государственного университета. Вся трудовая деятельность Баира Сыдыповича связана с Институтом катализа, где он прошел путь от стажера-исследователя до заместителя директора по научной работе. В 1978 году им была защищена кандидатская диссертация, а в 1991 году – докторская диссертация на тему “Применение релаксационных методов в гетерогенном катализе”. В последние годы Б.С. Бальжинимаев возглавлял лабораторию исследования и испытания новых материалов в катализе.
Б.С. Бальжинимаев является автором более 200 статей в рецензируемых журналах и более 50 патентов. Им развито перспективное направление – исследование механизмов каталити-ческих реакций с помощью кинетических релаксационных и изотопно-динамических методов в сочетании с различными физическими методами. Разработаны и применены теоретические и экспериментальные подходы для ряда конкретных реакций парциального окисления углеводородов, включая окисление C-H связей в низших парафинах, окисление диоксида серы, селективное восстановление оксидов азота, эпоксидирование этилена молекулярным кислородом. В некоторых случаях на атомно-молекулярном уровне установлена количественная взаимосвязь между скоростью реакции, составом адсорбированного слоя и характеристиками состояния катализатора. Разработаны и реализованы подходы к молекулярному дизайну высокоэффективных катализаторов синтеза серной кислоты, оксида этилена.
В последнее время научная деятельность Б.С. Бальжинимаева была сконцентрирована на поисковых работах, связанных с разработкой эффективных катализаторов на основе новых материалов, таких как синтетический углерод, стекловолокна, нанодисперсные металлы. В результате найдены новые композиты, проявляющие высокие активность и селективность в различных реакциях. Среди них особое место занимают катализаторы для эпоксидирования легких олефинов, очистки отходящих газов промышленности от вредных соединений, селективного гидрирования ацетиленовых углеводородов, новых процессов синтеза винилхлорида, процесса получения легких олефинов из природного газа.
Были коммерциализированы катализаторы производства серной кислоты и очистки газовых выбросов от вредных веществ.
Являясь заместителем директора Института по научной работе с 1997 по 2015 годы, Б.С. Бальжинимаев проводил активную научно-техническую политику с ведущими химическими и нефтехимическими компаниями США, Европы, Японии, Южной Кореи и ЮАР. В результате заключено и реализовано несколько десятков исследовательских и лицензионных контрактов. Под его руководством выполнялись государственные контракты, направленные на создание эффективных производств в сфере реальной экономики страны.
Б.С. Бальжинимаев вел большую научно-организационную работу, связанную с сотрудничеством в области катализа с зарубежными исследовательскими центрами и компаниями химической ориентации, являлся координатором с российской стороны в ряде международных проектов со странами Европейского Сообщества, США и НАТО, входил в состав редколлегии международного журнала Journal of Energy Chemistry, был заместителем главного редактора журнала “Катализ в промышленности”, являлся рецензентом в международных журналах ACS Catalysis, ACS Sustainable Chemistry & Engineering, RSC Advances, ChemCatChem, Applied Catalysis A и B, Journal of Catalysis, Journal of Energy Chemistry. Принимал активное участие в подготовке научных кадров. Под его руководством были защищены восемь кандидатских диссертаций.
Б.С. Бальжинимаев являлся членом Ученого совета Института катализа, заместителем председателя диссертационного совета, экспертом научных проектов для Российской академии наук, членом Научного совета по катализу РАН.
Коллеги и ученики Баира Сыдыповича будут помнить о нем как о человеке с безграничной жизненной энергией, наполненном новыми перспективными идеями и посвятившем всю свою жизнь служению науке.
Памяти Валерия Кузьмича ДУПЛЯКИНА
(1939 – 2020 гг.)

28 октября 2020 года ушел из жизни Валерий Кузьмич Дуплякин – известный ученый, доктор химических наук, дважды лауреат Премии правительства Российской Федерации в области науки и техники, Заслуженный ветеран Сибирского отделения РАН, основатель и первый руководитель Омского филиала ИК СО РАН.
В.К. Дуплякин – специалист в области каталитических превращений углеводородов, автор и соавтор более 240 научных публикаций, в том числе 44 изобретений. Его работы широко известны в России и за рубежом.
Работу в Сибирском отделении РАН В.К. Дуплякин начал в 1978 году в качестве организатора одного из первых академических научных подразделений в г. Омске – отдела Института катализа АН СССР, который в 1991 г. был реорганизован в Омский филиал ИК СО
Под руководством В.К. Дуплякина был создан научный коллектив специалистов, научно-экспериментальная и опытно-производственная база, обеспечивающие выполнение научных исследований в области нефтепереработки и нефтехимии, а также прикладных разработок в интересах, прежде всего, крупнейшего в мире комплекса омских химических предприятий.
Успешное развитие работ в научном коллективе под его руководством привело к получению принципиально важных результатов для формирования научных представлений о конструировании катализаторов. Существенность достигнутых научных результатов нашла отражение в создании промышленных катализаторов для базовых процессов нефтепереработки (крекинг и риформинг), которые по ряду показателей превышают мировой уровень, успешно производятся и эксплуатируются на нефтеперерабатывающих заводах страны.
В.К. Дуплякин в составе коллектива авторов дважды был удостоен премии Правительства РФ в области науки и техники: в 1996 году за работу «Разработка, внедрение в производство и использование эффективных катализаторов крекинга», в 2019 году за работу «Разработка новых импортозамещающих техно-логий производства катализаторов риформинга и их промышленное освоение на нефтеперерабатывающих заводах РФ».
За свою научную и научно-организационную деятельность В.К. Дуплякин был награжден орденом “Знак почета”, медалью ордена “За заслуги перед отечеством” II степени, неоднократно награждался почетными грамотами Президиума Сибирского отделения РАН.
Валерия Кузьмича Дуплякина отличал высокий профессионализм, преданность науке, доброжелательность. Благодаря этим качествам он пользовался заслуженным уважением и авторитетом среди коллег по работе и учеников, которые сохранят в своей памяти самые теплые воспоминания о Валерии Кузьмиче – учителе, ученом и замечательном человеке.
Памяти Романа Алексеевича БУЯНОВА
(1927 – 2020 гг.)

4 декабря 2020 года ушел из жизни член-корреспондент РАН, доктор химических наук, профессор Роман Алексеевич Буянов.
Р.А. Буянов начал свою трудовую деятельность на Чирчикском электрохимическом комбинате, позднее руководил сооружением ряда крупных промышленных объектов, в том числе, первого в Средней Азии завода сухого льда, крупной ТЭЦ, цеха азотной кислоты. В 1958-1961 гг. работал в Объединенном институте ядерных исследований в Дубне.
В 1960 г. за разработку и промышленное освоение технологии получения дейтерия методом ректификации жидкого водорода Р.А. Буянову присвоено звание Лауреата Ленинской премии.
В 1961 г. Роман Алексеевич защитил кандидатскую диссертацию и был приглашен Г.К. Боресковым в Институт катализа в СО АН СССР на должность его заместителя. С августа 1961 г. совмещал три должности: заместителя директора по науке, главного инженера и заведующего лабораторией. До 1964 г. руководил строительством Института, организацией его инфраструктуры и всех его служб. Одновременно участвовал в решении ряда вопросов, связанных с организацией СО АН СССР.
В 1967 г. за участие в создании Сибирского отделения АН СССР и развитие науки в Сибири награжден орденом Трудового Красного знамени.
В 1972 г. защитил докторскую диссертацию. В 1976 г. ему присвоено звание профессора, а в 1977 г. – почетное звание Заслуженный деятель науки РСФСР. В 1981 году избран членом-корреспондентом АН СССР. В 1982 г. награжден вторым орденом Трудового Красного знамени, в 1987 г. – орденом Октябрьской революции.
Научно-исследовательская деятельность Р.А. Буянова связана с развитием научных основ приготовления катализаторов и углерод-минеральных носителей, в том числе с применением механохимии. Исследуя причины дезактивации и разрушения промышленных катализаторов, дал им научную классификацию и раскрыл механизм “карбидного цикла” при зауглероживании катализаторов. При непосредственном участии Романа Алексеевича разработаны и освоены в промышленности катализаторы получения мономеров синтетического каучука, катализаторы сероочистки по методу Клауса, углерод-минеральные носители и катализаторы получения винилхлорида, носители на основе оксидов алюминия, катализаторы дегидрирования и др.
Р.А. Буяновым была разработана фундаментальная теория магнитного действия катализаторов в низкотемпературной конверсии орто-водорода в пара-водород. Эти работы завершились созданием промышленного производства жидкого пара-водорода – ракетного топлива, на котором совершен полет космического корабля “Буран”.
В течение 10 лет Р.А. Буянов был руководителем Координационного совета, представителем СССР в Совете уполномоченных стран СЭВ по созданию и освоению новых промышленных катализаторов.
В 1996 году перешел на должность советника РАН. По-прежнему активно участвовал в решении научных задач лаборатории дегидрирования, являлся членом научных и ученых советов. Был главным редактором журнала “Известия Сибирского отделения. Серия химических наук”, заместителем главного редактора международного журнала “Химия в интересах устойчивого развития”, членом редколлегий журналов “Катализ в промышленности” и “Кинетика и катализ”.
Роман Алексеевич прожил яркую жизнь. Это был удивительный человек, с огромным жизненным опытом, обладающий творческим мышлением. Через годы он сумел пронести такие ценности, как патриотизм, честь, долг, семья. Для многих стал другом, мудрым наставником, терпеливым учителем. Уход из жизни Р.А. Буянова – невосполнимая потеря.