23 июля исполнилось 65 лет Александру Степановичу Носкову – доктору технических наук,
профессору, крупному специалисту в области технологии каталитических процессов и математического
моделирования каталитических
реакторов.
23 июля исполнилось 65 лет Александру Степановичу Носкову – доктору технических наук,
профессору, крупному специалисту в области технологии каталитических процессов и математического
моделирования каталитических
реакторов.
В 1976 году А.С. Носков окончил Томский политехнический университет (ТПУ) и в течение двух лет работал в отраслевой лаборатории Миннефтехимпрома СССР при ТПУ. Дальнейшая трудовая деятельность Александра Степановича связана с Институтом катализа им. Г.К. Борескова СО РАН, где он прошел путь от аспиранта до заместителя директора по науке (с 1995 г. по настоящее время).
С 1985 г. по 1992 г. А.С. Носков возглавлял научную программу работ Сибирского отделения РАН по каталитическим методам очистки от токсичных примесей (оксиды азота, углеводороды), которая завершилась созданием промышленного производства установок очистки газов. Было построено и введено в эксплуатацию более 20 установок на химических, машиностроительных предприятиях и заводах военно-промышленного комплекса.
Работы А.С. Носкова по использованию методов вычислительной гидродинамики при разработке и эксплуатации каталитических реакторов позволили разработать оптимальные способы формирования зернистых структур в неподвижных слоях катализатора, показать возможность повышения производительности промышленных реакторов на 12-15% и обеспечить применение этих методов более чем в 60 агрегатах в азотной промышленности и нефтепереработке.
Под руководством А.С. Носкова разработаны научные основы селективного окисления аммиака в закись азота и впервые в мире создан в пилотном масштабе реактор получения закиси азота путем окисления аммиака в кипящем слое катализатора.
В период с 2003 по 2006 гг. А.С. Носков обеспечил в качестве исполнительного директора проекта успешное выполнение важнейшего государственного инновационного проекта по созданию и промышленному освоению нового поколения катализаторов получения моторных топлив. Экономический эффект от выполнения данного проекта превысил 10 млрд. рублей. В настоящее время А.С. Носков осуществляет научное руководство созданием крупнейшего в России производства катализаторов в г. Омске, которое обеспечивает импортозамещение базовых катализаторов глубокой переработки нефтяного сырья.
В 2009-2013 гг. под руководством А.С. Носкова был успешно реализован научный проект по разработке и освоению нового поколения теплогенерирующих систем на основе каталитического сжигания твердых топлив. К настоящему времени создано и эксплуатируется в промышленности пять таких систем, позволивших снизить расход твердого топлива в 2-2,5 раза.
Деятельность А.С. Носкова обеспечила успешное выполнение ряда крупных научно-технических проектов. К числу таких проектов относится региональная программа «Экология городов Сибири», которая завершилась созданием промышленного производства установок каталитической очистки газов и вводом в эксплуатацию нескольких десятков установок на предприятиях Новосибирска, Кемерово, Омска, Бийска.
Под руководством А.С. Носкова с 2007 г. выполнено 20 государственных контрактов в рамках Федеральных целевых программ «Национальная технологическая база», «Исследования и разработки по приоритетным направлениям развития научно-технологического комплекса России», «Кадры».
Александр Степанович внес значительный вклад в формирование научно-образовательного комплекса – в 1998 г. он основал и по 2006 г. возглавлял кафедру «Инженерные проблемы экологии» Новосибирского государственного технического университета. За годы существования кафедры на ней подготовлено более 200 инженеров-экологов, которые востребованы предприятиями ТЭК, машиностроения и научной сферы.
А.С. Носков является автором и соавтором более чем 600 научных публикаций в ведущих российских и международных журналах, включая 3 монографии, более 100 российских и зарубежных патентов.
Александр Степанович Носков является признанным экспертом в области химической технологии и входит в состав ряда Научных Советов Российской академии наук: по катализу, по теоретическим основам химической технологии, по ископаемому сырью, участвует в работе редакционных коллегий нескольких российских журналов и международного журнала “Reviews in Chemical Engineering”. С 2001 г. под руководством А.С. Носкова проводятся традиционные Международные конференции по химическим реакторам (в Финляндии, Германии, Греции, на Мальте, в Австрии), которые объединяют ведущих российских и зарубежных ученых в области теоретических основ создания каталитических реакторов и процессов.
С 2006 г. А.С. Носков входит в состав экспертного совета Высшей аттестационной комиссии России. Заслуги А.С. Носкова отмечены государственной (Орден Почета, 2007 г.) и региональной (медаль за особый вклад в развитие Кузбасса, 2009 г.) наградами.
Научный совет по катализу ОХНМ РАН и редакция Каталитического бюллетеня сердечно поздравляют Александра Степановича с юбилеем, желают ему крепкого здоровья и новых творческих достижений!
16-20 апреля 2018 г., Лион, Франция
Научно-технологический симпозиум «Нефтепереработка: катализаторы и гидропроцессы» проводится раз в два года и признается мировым научным и производственным сообществом как мероприятие высокого международного уровня. Симпозиум является практически единственным европейским форумом по гидропроцессам и каталитическим гидрогенизационным технологиям нефтепереработки. В 2018 году III Научно-технологический симпозиум «Нефтепереработка: катализаторы и гидропроцессы» прошел с 16 по 20 апреля в Лионе (Франция).

Выбор места проведения симпозиума связан с тем, что Лион является технологически ориентированным городом. Это третий по величине город Франции и главный центр химической, фармацевтической и биотехнологической промышленности. Наличие большого числа университетов, исследовательских компаний, а также крупных промышленных комплексов создает активную творческую атмосферу. Лион по праву называют европейской столицей катализа. Это город развитой науки, именно поэтому он привлекает ученых со всего мира для участия в многочисленных научных мероприятиях, которые обычно проводятся в Лионе.
Соорганизаторами симпозиума стали Институт катализа СО РАН им. Г.К. Борескова (Новосибирск, Россия), Институт катализа и защиты окружающей среды в Лионе (IRCELYON), Институт нефти (IFPEN) (Лион, Франция), Институт проблем переработки углеводородов СО РАН (Омск, Россия), ПАО «Газпром нефть» (Санкт-Петербург, Россия). Председателями симпозиума традиционно выступили профессор Жильбер Фрома (Бельгия) и академик Валентин Пармон (ИК СО РАН, Новосибирск, Россия). Экспертный Совет возглавили д.т.н., профессор Александр Носков (ИК СО РАН, Новосибирск, Россия), доктор Катрин Пинель, директор Института катализа и защиты окружающей среды в Лионе (IRCELYON), и доктор Люк Ноугиер (Институт нефти (IFPEN), Лион, Франция).

Традиционно в работе симпозиума принимают участие лидеры в области развития технологий гидрогенизационных процессов нефтепереработки, представляющие как ведущие университеты мира, так и крупные нефтехимические компании. В работе предыдущих симпозиумов, прошедших в Санкт-Петербурге и Белграде, приняли участие около 300 специалистов из 25 стран мира. Труды этих симпозиумов были опубликованы в журнале «Катализ в промышленности». В 2018 году организаторы получили приглашение от главного редактора журнала Catalysis Today (издательство Эльзевир) профессора Джеймса Спивея на публикацию статей симпозиума в специальном выпуске журнала. Журнал Catalysis Today имеет высокий импакт-фактор и мировую известность. Это приглашение свидетельствует о признании высокого уровня проведения симпозиума.
Научная программа симпозиума охватывала следующие направления:
В работе III Научно-технологического симпозиума приняли участие 120 специалистов в области нефтехимии из 17 стран мира, среди которых были представители России, Франции, Канады, США, Германии, Китая, Индии, Мексики, Кувейта, Южной Кореи, Испании, Нидерландов, Греции, Австрии, Дании, ОАЭ, Италии. В рамках научной программы было представлено 7 пленарных и 6 ключевых лекций специалистов мирового уровня, 46 устных и 35 стендовых докладов. Большой интерес вызвал круглый стол «Основные тенденции и долгосрочные перспективы развития катализаторов гидропроцессов и технологий их производства», инициированный Генеральным спонсором симпозиума, компанией ПАО «Газпром нефть», который прошел с привлечением экспертов из ICRELYON, IFPEN, UOP и академических организаций.
 Пленарную сессию симпозиума открыл профессорЖан-Пьер Жильсон, директор Лаборатории катализа и спектрохимии Университета Кана (Франция), консультант Даляньского университета (Китай) по международным академическим и
промышленным связям. Жан-Пьер Жильсон обладает большим опытом работы в ведущих мировых нефтехимических компаниях, среди которых Grace Davison, UOP, Shell, Total. Он известен как крупный специалист в области разработок и лицензирования технологий, а также как эксперт в маркетинговых исследованиях рынков нефтехимической продукции. Профессор Жильсон является создателем
трех химических процессов, реализованных в промышленности. Его лекция представляла собой краткий обзор перспектив применения цеолитных катализаторов в нефтепереработке с акцентом на гидропроцессы. Продемонстрированные в лекции результаты представляют большой теоретический и практический интерес и создают основу для разработки цеолитных катализаторов гидрогенизационных процессов в
нефтепереработке. Профессор Жан-Пьер Жильсон является научным руководителем российской инновационной
компании «ЭТБ каталитические технологии», занимающейся разработкой новых катализаторов.
Пленарную сессию симпозиума открыл профессорЖан-Пьер Жильсон, директор Лаборатории катализа и спектрохимии Университета Кана (Франция), консультант Даляньского университета (Китай) по международным академическим и
промышленным связям. Жан-Пьер Жильсон обладает большим опытом работы в ведущих мировых нефтехимических компаниях, среди которых Grace Davison, UOP, Shell, Total. Он известен как крупный специалист в области разработок и лицензирования технологий, а также как эксперт в маркетинговых исследованиях рынков нефтехимической продукции. Профессор Жильсон является создателем
трех химических процессов, реализованных в промышленности. Его лекция представляла собой краткий обзор перспектив применения цеолитных катализаторов в нефтепереработке с акцентом на гидропроцессы. Продемонстрированные в лекции результаты представляют большой теоретический и практический интерес и создают основу для разработки цеолитных катализаторов гидрогенизационных процессов в
нефтепереработке. Профессор Жан-Пьер Жильсон является научным руководителем российской инновационной
компании «ЭТБ каталитические технологии», занимающейся разработкой новых катализаторов.
 Доктор Бенуа Келсе, который представил следующую пленарную лекцию, работает в Отделе проектирования и моделирования процессов Французского института нефти (IFPEN) в Лионе. Его научные интересы лежат в области технологической инженерии, химических реакций, кинетики, реакционной кинетики, катализа, кинетического моделирования, нефтехимии, дизельных двигателей. В своей лекции «Кинетическое моделирование процессов гидрокрекинга» (Kinetic modeling of hydrocracking processes) доктор Келсе продемонстрировал глубокие знания в области гидрогенизационых процессов и опыт моделирования процессов гидропереработки, создания кинетических моделей, хемометрики, диагностики сбоев в сложных процессах. Лекция была посвящена вопросам переработки тяжелой нефти и дистиллятов (керосин и дизельное топливо) путем каталитического гидрокрекинга. Были представлены результаты моделирования процесса гидрокрекинга. Доктор Келсе изложил основные результаты, полученные для нескольких типов моделей, а также представил
новые тенденции в этой области, объединив технологии Big Data с методами кинетического моделирования.
Доктор Бенуа Келсе, который представил следующую пленарную лекцию, работает в Отделе проектирования и моделирования процессов Французского института нефти (IFPEN) в Лионе. Его научные интересы лежат в области технологической инженерии, химических реакций, кинетики, реакционной кинетики, катализа, кинетического моделирования, нефтехимии, дизельных двигателей. В своей лекции «Кинетическое моделирование процессов гидрокрекинга» (Kinetic modeling of hydrocracking processes) доктор Келсе продемонстрировал глубокие знания в области гидрогенизационых процессов и опыт моделирования процессов гидропереработки, создания кинетических моделей, хемометрики, диагностики сбоев в сложных процессах. Лекция была посвящена вопросам переработки тяжелой нефти и дистиллятов (керосин и дизельное топливо) путем каталитического гидрокрекинга. Были представлены результаты моделирования процесса гидрокрекинга. Доктор Келсе изложил основные результаты, полученные для нескольких типов моделей, а также представил
новые тенденции в этой области, объединив технологии Big Data с методами кинетического моделирования.
 Профессор
Педро Перейра-Алмао, представлявший Университет Калгари (Канада), выступил с лекцией «Пути обновления каталитической отрасли: что работает и что нет» (In the path for catalytic field upgrading: what works and what doesn’t). Было отмечено, что со времен первого полевого испытания технологии вакуумной паровой переработки (акваконверсии), проведенного в 1996 году, разработка каталитических технологий для переработки тяжелой нефти стала высокоэффективным направлением. Развитие каталитических технологий – это путь модернизации нефтеперерабатывающих компаний, который позволяет снизить уровень воздействия на окружающую среду и риски роста модульной добычи нефти, дает возможность полностью использовать нефтеперерабатывающие заводы с глубокой конверсией, минимизирует твердые побочные продукты и т.п. Было кратко описано расширение области применения каталитических методов в процессах гидрообработки и парообразования нефракционированных сверхтяжелых масел. Представлены подходы с использованием новых ультрадисперсных катализаторов, нанокатализаторов и катализаторов с неподвижным слоем, работающих с нефракционированными сверхтяжелыми маслами и более адаптированных к процессам модернизации.
Профессор
Педро Перейра-Алмао, представлявший Университет Калгари (Канада), выступил с лекцией «Пути обновления каталитической отрасли: что работает и что нет» (In the path for catalytic field upgrading: what works and what doesn’t). Было отмечено, что со времен первого полевого испытания технологии вакуумной паровой переработки (акваконверсии), проведенного в 1996 году, разработка каталитических технологий для переработки тяжелой нефти стала высокоэффективным направлением. Развитие каталитических технологий – это путь модернизации нефтеперерабатывающих компаний, который позволяет снизить уровень воздействия на окружающую среду и риски роста модульной добычи нефти, дает возможность полностью использовать нефтеперерабатывающие заводы с глубокой конверсией, минимизирует твердые побочные продукты и т.п. Было кратко описано расширение области применения каталитических методов в процессах гидрообработки и парообразования нефракционированных сверхтяжелых масел. Представлены подходы с использованием новых ультрадисперсных катализаторов, нанокатализаторов и катализаторов с неподвижным слоем, работающих с нефракционированными сверхтяжелыми маслами и более адаптированных к процессам модернизации.
 Пленарную лекцию представил д.х.н., заместитель директора по научной работе Института катализа СО РАН Олег Николаевич Мартьянов. Тема его лекции – Стабильность и эволюция систем тяжелой нефти, изучаемых с помощью передовых методов in situ (The stability and evolution of heavy oil systems studied via advanced methods in situ). Современная нефтяная промышленность столкнулась с серьезным вызовом, который связан с необходимостью развития эффективных технологий добычи и последующей глубокой переработки тяжелых нефтей и их компонентов. Проблемы использования такого углеводородного сырья
связаны с дороговизной его извлечения и транспортировки, а также с принципиальными трудностями, которые возникают при переработке
тяжелых нефтей с применением традиционных каталитических технологий. Одной из наиболее труднопреодолимых проблем является образование отложений в технологическом оборудовании при транспортировке и переработке тяжелых нефтей, что приводит к неэффективному теплообмену, заметным потерям энергии и сырья в ходе технологического процесса, которые могут составлять до нескольких процентов энергетического запаса (эквивалента) сырой нефти. В лекции были проанализированы возможности современных методов, которые позволяют исследовать in situ фазовую стабильность и физико-химические превращения, происходящие в тяжелых
нефтях; изложены основные принципы применения методов в режиме in situ, рассмотрены возможности и ограничения каждого подхода. Особое внимание уделено методам визуализации процессов агрегации асфальтенов с использованием ИК-Фурье спектроскопии нарушенного внутреннего отражения и магнитно-резонансной томографии, а также методам малоуглового рентгеновского и нейтронного рассеяния, электронному магнитному резонансу, электронной и оптической микроскопии. Обсуждаемые в лекции методы исследования позволяют получать взаимодополняющую информацию о свойствах и поведении углеводородных дисперсных систем на различных пространственных и временных масштабах, начиная с вращательной подвижности молекул асфальтенов и динамики их локального окружения с характерным временем ~10-10 сек, эволюции размера и формы их агрегатов, до визуализации процессов
агрегации и формирования отложений в нефтях и блендах, на масштабах от нескольких единиц до тысяч микрометров с характерным
временем от секунд до сотен часов. Описанные подходы могут эффективно применяться в широком диапазоне температур и давлений, а также при введении в систему специальных химических веществ, которые влияют на стабильность тяжелых нефтей.
Пленарную лекцию представил д.х.н., заместитель директора по научной работе Института катализа СО РАН Олег Николаевич Мартьянов. Тема его лекции – Стабильность и эволюция систем тяжелой нефти, изучаемых с помощью передовых методов in situ (The stability and evolution of heavy oil systems studied via advanced methods in situ). Современная нефтяная промышленность столкнулась с серьезным вызовом, который связан с необходимостью развития эффективных технологий добычи и последующей глубокой переработки тяжелых нефтей и их компонентов. Проблемы использования такого углеводородного сырья
связаны с дороговизной его извлечения и транспортировки, а также с принципиальными трудностями, которые возникают при переработке
тяжелых нефтей с применением традиционных каталитических технологий. Одной из наиболее труднопреодолимых проблем является образование отложений в технологическом оборудовании при транспортировке и переработке тяжелых нефтей, что приводит к неэффективному теплообмену, заметным потерям энергии и сырья в ходе технологического процесса, которые могут составлять до нескольких процентов энергетического запаса (эквивалента) сырой нефти. В лекции были проанализированы возможности современных методов, которые позволяют исследовать in situ фазовую стабильность и физико-химические превращения, происходящие в тяжелых
нефтях; изложены основные принципы применения методов в режиме in situ, рассмотрены возможности и ограничения каждого подхода. Особое внимание уделено методам визуализации процессов агрегации асфальтенов с использованием ИК-Фурье спектроскопии нарушенного внутреннего отражения и магнитно-резонансной томографии, а также методам малоуглового рентгеновского и нейтронного рассеяния, электронному магнитному резонансу, электронной и оптической микроскопии. Обсуждаемые в лекции методы исследования позволяют получать взаимодополняющую информацию о свойствах и поведении углеводородных дисперсных систем на различных пространственных и временных масштабах, начиная с вращательной подвижности молекул асфальтенов и динамики их локального окружения с характерным временем ~10-10 сек, эволюции размера и формы их агрегатов, до визуализации процессов
агрегации и формирования отложений в нефтях и блендах, на масштабах от нескольких единиц до тысяч микрометров с характерным
временем от секунд до сотен часов. Описанные подходы могут эффективно применяться в широком диапазоне температур и давлений, а также при введении в систему специальных химических веществ, которые влияют на стабильность тяжелых нефтей.
 Следующую пленарную лекцию представил доктор Паскаль Райбауд из IFP Energies nouvelles (Лион, Франция): «Молекулярный взгляд на ключевые компоненты и функции катализаторов для гидропроцессов» (A molecular
view of the key components and functions of hydroprocessing catalysts). В лекции было отмечено, что процессы гидрообработки и гидрокрекинга
приобретают все большее значение для получения топлива из нефтяного сырья, которое становится сложнее перерабатывать. Эти процессы
включают в себя широкий спектр механизмов реакции, активируемых различными каталитическими материалами: сульфидами переходных
металлов, алюмосиликатами и т.п. Для содействия гидрогенолизу, гидрированию или крекингу необходимо активировать связи C-S, C-N, C-H
или C-C с помощью широкого спектра активных компонентов. Понимание этих процессов на молекулярном уровне представляет собой важный шаг для улучшения рационального проектирования этих катализаторов. Данная лекция проиллюстрировала, как современное квантовое моделирование, основанное на теории функционала плотности (DFT), может быть мощным подходом к решению проблемы разнообразия катализаторов гидрообработки и их многокомпонентного характера. С одной стороны, было показано, как функции гидрирования/гидрогенолиза активных центров сульфидов переходных металлов могут быть описаны с помощью DFT-моделирования.
Были выделены особенности атомного уровня активной фазы и взаимодействие сульфидированных активных центров с соответствующими
молекулами, обнаруженными при гидрообработке. Что касается функций крекинга и изомеризации, были представлены результаты теоретических исследований на кислотных центрах Бренстеда во взаимодействии с углеводородами на алюмосиликатных материалах, подходящих для гидрообработки, таких как объемные цеолиты, деалюминированные цеолиты, поверхности цеолита и аморфные диоксиды кремния-оксиды алюминия. Было показано, как DFT-расчеты помогают интерпретировать экспериментальные наблюдения на этих сложных катализаторах.
Следующую пленарную лекцию представил доктор Паскаль Райбауд из IFP Energies nouvelles (Лион, Франция): «Молекулярный взгляд на ключевые компоненты и функции катализаторов для гидропроцессов» (A molecular
view of the key components and functions of hydroprocessing catalysts). В лекции было отмечено, что процессы гидрообработки и гидрокрекинга
приобретают все большее значение для получения топлива из нефтяного сырья, которое становится сложнее перерабатывать. Эти процессы
включают в себя широкий спектр механизмов реакции, активируемых различными каталитическими материалами: сульфидами переходных
металлов, алюмосиликатами и т.п. Для содействия гидрогенолизу, гидрированию или крекингу необходимо активировать связи C-S, C-N, C-H
или C-C с помощью широкого спектра активных компонентов. Понимание этих процессов на молекулярном уровне представляет собой важный шаг для улучшения рационального проектирования этих катализаторов. Данная лекция проиллюстрировала, как современное квантовое моделирование, основанное на теории функционала плотности (DFT), может быть мощным подходом к решению проблемы разнообразия катализаторов гидрообработки и их многокомпонентного характера. С одной стороны, было показано, как функции гидрирования/гидрогенолиза активных центров сульфидов переходных металлов могут быть описаны с помощью DFT-моделирования.
Были выделены особенности атомного уровня активной фазы и взаимодействие сульфидированных активных центров с соответствующими
молекулами, обнаруженными при гидрообработке. Что касается функций крекинга и изомеризации, были представлены результаты теоретических исследований на кислотных центрах Бренстеда во взаимодействии с углеводородами на алюмосиликатных материалах, подходящих для гидрообработки, таких как объемные цеолиты, деалюминированные цеолиты, поверхности цеолита и аморфные диоксиды кремния-оксиды алюминия. Было показано, как DFT-расчеты помогают интерпретировать экспериментальные наблюдения на этих сложных катализаторах.
 Далее в научной программе была представлена лекция «Иерархические цеолиты для конверсии углеводородов» (Hierarchical zeolites for hydrocarbon conversion) профессора Эмиля Дж. М. Хэнсена изТехнологического университета
Эйндховена (Нидерланды). Цеолиты имеют существенное значение для катализа. Их широкое применение может быть связано с различиями
размеров пор, большой площадью поверхности, высокой химической и термической стабильностью, а также с наличием более 220 топологий,
которые могут быть нацелены на соответствующий синтез. В лекции было рассказано о многомасштабных спектроскопических, микроскопических и вычислительных подходах, используемых не только для исследования процесса формирования цеолита, но и для разработки дешевого процесса темплатного синтеза иерархических цеолитов. В лекции был подробно описан синтез нанолистов ZSM-5
толщиной 2 нм, применяемых в ряде важных реакций гидроизомеризации, гидрокрекинга и депарафинизации.
Далее в научной программе была представлена лекция «Иерархические цеолиты для конверсии углеводородов» (Hierarchical zeolites for hydrocarbon conversion) профессора Эмиля Дж. М. Хэнсена изТехнологического университета
Эйндховена (Нидерланды). Цеолиты имеют существенное значение для катализа. Их широкое применение может быть связано с различиями
размеров пор, большой площадью поверхности, высокой химической и термической стабильностью, а также с наличием более 220 топологий,
которые могут быть нацелены на соответствующий синтез. В лекции было рассказано о многомасштабных спектроскопических, микроскопических и вычислительных подходах, используемых не только для исследования процесса формирования цеолита, но и для разработки дешевого процесса темплатного синтеза иерархических цеолитов. В лекции был подробно описан синтез нанолистов ZSM-5
толщиной 2 нм, применяемых в ряде важных реакций гидроизомеризации, гидрокрекинга и депарафинизации.
 Завершением пленарной сессии стало выступление профессора Берта М. Векхайсена из Университета Утрехта (Нидерланды) с
лекцией «Операндо-спектроскопия в катализе на цеолитах: от реактора до активного центра» (Operando
spectroscopy of zeolite-based catalysis: from reactor to active site). Докладчиком было отмечено, что цеолиты играют важную роль в существующих и разрабатываемых каталитических процессах, поскольку они сочетают высокую активность и стабильность с вариативностью по форме. Часто в промышленных операциях кристаллы цеолита укладываются в объемные экструдаты миллиметрового размера или сферические частицы микронного размера. Эти экструдаты и частицы содержат материалы, такие как оксид алюминия, диоксид кремния и глинистые минералы, которые улучшают термические, структурные и каталитические свойства полученных каталитических объектов. Понимание эволюции и распределения этих компонентов имеет решающее значение для их оптимальной работы. Был представлен обзор современных методов определения характеристик, доступных для выявления неоднородностей в цеолитных катализаторах. Особый акцент был сделан на явлениях, связанных с пространственно-временными характеристиками разного масштаба: на уровне каталитических реакторов (от мм до см), корпусов катализатора (от мкм до мм), зерен катализатора (от нм до мкм) и активных центров (от Å до нм). Были обсуждены спектроскопические методы визуализации пространственных неоднородностей в пространстве и времени; особенности оптических и синхротронных методов, их возможности в обеспечении пространственного разрешения (одномерное и двумерное химическое изображение) и профилировании глубины (трехмерное химическое изображение).
Завершением пленарной сессии стало выступление профессора Берта М. Векхайсена из Университета Утрехта (Нидерланды) с
лекцией «Операндо-спектроскопия в катализе на цеолитах: от реактора до активного центра» (Operando
spectroscopy of zeolite-based catalysis: from reactor to active site). Докладчиком было отмечено, что цеолиты играют важную роль в существующих и разрабатываемых каталитических процессах, поскольку они сочетают высокую активность и стабильность с вариативностью по форме. Часто в промышленных операциях кристаллы цеолита укладываются в объемные экструдаты миллиметрового размера или сферические частицы микронного размера. Эти экструдаты и частицы содержат материалы, такие как оксид алюминия, диоксид кремния и глинистые минералы, которые улучшают термические, структурные и каталитические свойства полученных каталитических объектов. Понимание эволюции и распределения этих компонентов имеет решающее значение для их оптимальной работы. Был представлен обзор современных методов определения характеристик, доступных для выявления неоднородностей в цеолитных катализаторах. Особый акцент был сделан на явлениях, связанных с пространственно-временными характеристиками разного масштаба: на уровне каталитических реакторов (от мм до см), корпусов катализатора (от мкм до мм), зерен катализатора (от нм до мкм) и активных центров (от Å до нм). Были обсуждены спектроскопические методы визуализации пространственных неоднородностей в пространстве и времени; особенности оптических и синхротронных методов, их возможности в обеспечении пространственного разрешения (одномерное и двумерное химическое изображение) и профилировании глубины (трехмерное химическое изображение).
Также в ходе симпозиума были представлены следующие ключевые лекции:
Профессор Игорь Коптюг Международный томографический центр СО РАН, Новосибирск, Россия «ЯМР-спектроскопия и изображение процессов преобразования углеводородов» (NMR spectroscopy and imaging of hydrocarbon conversion processes)
Профессор Каке Чжу Восточно-Китайский университет науки и технологий, Шанхай, Китай «Цеолиты с иерархической пористой структурой для производства углеводородов» (Zeolites with hierarchical pore structure for hydrocarbon feedstock production)
Доктор Леон С.А. ван ден Оетелаар Albemarle Catalysts Company BV, Амстердам, Нидерланды «STEM-EDX характеризация сульфидных катализаторов для гидропроцессов» (STEM-EDX characterization of sulfidic hydroprocessing catalysts)
Доктор Сесиль Сиеви CanmetENERGY, Девон, Альберта, Канада «Улучшение и переработка канадской нефти битуминозных песков – исследование CanmetENERGY» (Upgrading and refining of Canadian oil sands bitumen – research at CanmetENERGY, Natural Resources Canada)
Доктор Педро Кастано University of the Basque Country, Бильбао, Испания «Оценка каталитических свойств с помощью FT-ICR при гидропереработке тяжелой нефти» (Assessing the catalytic performance during heavy-oil hydroprocessing by FT-ICR)
Доктор Анил Кумар Синха CSIR-Indian Institute of Petroleum, Дехрадун, ИндияAcSIR-Academy of Scientific and Innovative Research, Ченнай, Индия «Селективная полиароматическая сатурация и размыкание кольца при гидропереработке легкого рециклового газойля на сульфидированном катализаторе Ni-Mo/ SIO2-AL2O3 (Selective polyaromatics saturation and ring opening during hydroprocessing of light cycle oil over sulfided Ni-Mo/SiO2-Al2O3 catalyst)

Круглый Стол «Основные тенденции и долгосрочные перспективы развития катализаторов гидропроцессов и технологий их производства», вызвавший живой интерес аудитории, был организован по инициативе Генерального спонсора симпозиума, ПАО «Газпром нефть» (Санкт-Петербург). Модератором и председателем Круглого стола выступил профессор Жилбер Фрома (Бельгия), сопредседателем – профессор Андрей Загоруйко (Институт катализа СО РАН, Россия). После яркого вступительного доклада профессора Фрома, в котором он обозначил задачи брифинга и отразил важность обозначенных тем, прозвучали выступления участников – как представителей университетов, так и крупных промышленных компаний. С краткими докладами выступили профессор Абдулазим Марафи (Кувейтский институт научных исследований), доктор Герхард Пирнгрубер (Институт нефти-IFPEN, Солаиз, Франция), профессор Хорхе Анчейта (Мексиканский нефтяной институт, Мехико). В их презентациях была представлена информация о долгосрочных перспективах и основных тенденциях развития каталитических процессов нефтепереработки, а также прорывных направлениях по созданию эффективных технологий производства катализаторов. Большое внимание было уделено вопросам гидрокрекинга тяжелого углеводородного сырья. Последовавшая активная и продолжительная дискуссия продемонстрировала интерес аудитории к затронутым проблемам. В ярких дебатах участвовали Генеральный директор компании «Природные ресурсы Канады», доктор Сисель Селве; пленарный лектор, профессор Жан-Пьер Жильсон (Франция); научный сотрудник Кувейтского института научных исследований, доктор Халида Ал-Далама; пленарный лектор, профессор Педро Перейра-Алмао (Университет Калгари, Канада); профессор Андрей Загоруйко (Институт катализа СО РАН, Россия); профессор Сержио Фуентес (Национальный автономный университет Мексики); доктор Майкл Бродо-Кэмпбелл (UOP, США); руководитель направления по связям с научными и учебными учреждениями ПАО «Газпром нефть», доктор Д.О. Кондрашев и другие участники симпозиума. Значимость проведению брифинга придавало присутствие представителей инициатора мероприятия, руководства ПАО «Газпром нефть»: начальника Департамента развития нефтепереработки и нефтехимии О.С. Ведерникова и начальника Управления научно-технического развития А.В. Клейменова.
Итоги проведения Круглого стола выразились в формулировании основных технологических тенденций в развитии гидропроцессов нефтепереработки на долгосрочную перспективу. Были отмечены проблемные вопросы, такие как изменение качества перерабатываемой нефти в сторону утяжеления углеводородного состава; ухудшение качества смесевого сырья гидроочистки и гидрокрекинга ввиду вовлечения вторичных фракций, большего содержания серы, азота, металлов и коксообразующих компонентов; возможное дальнейшее ужесточение экологических требований к моторным топливам.
В качестве долгосрочных исследовательских проектов поискового типа, имеющих потенциал «прорывных» технологий в ближайшие годы, были выделены следующие направления:
 В рамках Симпозиума успешно прошла работа выставки компании SPECS (Берлин, Германия). Представитель компании, доктор Лиана
Сокасиу-Зиберт, выступила с презентационным докладом, в котором рассказала об
основных направлениях деятельности компании и ключевых аспектах своих исследовательских интересов. Ее научная работа сосредоточена на получении сведений об элементарных стадиях каталитических процессов на атомном и молекулярном уровнях с использованием
технологии NAP-XPS. Выставка компании SPECS вызвала большой интерес со стороны участников симпозиума.
В рамках Симпозиума успешно прошла работа выставки компании SPECS (Берлин, Германия). Представитель компании, доктор Лиана
Сокасиу-Зиберт, выступила с презентационным докладом, в котором рассказала об
основных направлениях деятельности компании и ключевых аспектах своих исследовательских интересов. Ее научная работа сосредоточена на получении сведений об элементарных стадиях каталитических процессов на атомном и молекулярном уровнях с использованием
технологии NAP-XPS. Выставка компании SPECS вызвала большой интерес со стороны участников симпозиума.
На заключительном заседании симпозиума было отмечена крайняя важность и актуальность проведения мероприятий в области разработки новых, более совершенных каталитических систем для процессов гидроочистки и гидрокрекинга, которые являются ключевыми процессами современной нефтепереработки. Актуальность исследований обусловлена необходимостью удовлетворять растущие экологические требования к качеству выпускаемых заводами нефтепродуктов. От участников были получены предложения о месте и времени проведения следующего симпозиума.

Материал подготовили
А.Н. Загоруйко, О.В. Климов, А.С. Носков,
Т.В. Замулина, С.С. Логунова
Фото: П.М. Елецкий
(Институт катализа СО РАН, Новосибирск)
Проект Института катализа СО РАН «ЦКП «Опытное производство катализаторов» был представлен в последних числах августа на выставке в рамках VI Международного форума технологического развития «Технопром-2018» в новосибирском Экспоцентре. Стенд Проекта демонстрировался в составе коллективной экспозиции Администрации Новосибирской области «Академгородок 2.0», занявшей центральную часть выставочного комплекса.

На стенде проекта развития Новосибирского научного центра.
На фото (слева направо):
председатель СО РАН, академик Валентин Николаевич Пармон,
помощник Президента РФ Андрей Александрович Фурсенко,
министр науки и высшего образования Михаил Михайлович Котюков,
врио губернатора НСО Андрей Александрович Травников,
директор ИК СО РАН, академик Валерий Иванович Бухтияров,
мэр наукограда «Кольцово» Николай Григорьевич Красников
В этом году Форум во многом был посвящен развитию Сибирской науки и технологий. В нем приняли участие руководители страны и региона на самом высоком уровне. Самым главным участником мероприятия стал Президент России Владимир Владимирович Путин. Он выступил на пленарном заседании «Наука как индустрия. Повестка 2024» 28 августа, а также посетил выставку Форума, на которой были представлены высокотехнологичные разработки и проекты.
В работе Форума приняли активное участие помощник Президента РФ Андрей Александрович Фурсенко, министр науки и высшего образования Михаил Михайлович Котюков, полпред Президента по СФО Сергей Иванович Меняйло, врио губернатора НСО Андрей Александрович Травников, Председатель СО РАН и научный руководитель ИК СО РАН Валентин Николаевич Пармон.

Директор ИК СО РАН, академик Валерий Иванович Бухтияров
рассказывает о проекте развития ННЦ президенту РАН,
академику Александру Михайловичу Сергееву
Проект Института «Центр коллективного пользования «Опытное производство катализаторов» высоким гостям Форума представили директор ИК СО РАН академик Валерий Иванович Бухтияров и заместитель директора по научной работе д.х.н. Вадим Анатольевич Яковлев.

Заместитель директора ИК СО РАН, д.х.н. Вадим Анатольевич Яковлев
делает презентацию проекта ИК СО РАН
«ЦКП « Опытное производство катализаторов»
В рамках данного проекта планируется к 2025 году создание новых производственных мощностей и инфраструктуры для осуществления масштабного перехода от прикладных разработок в области каталитических технологий к их промышленному внедрению при производстве моторных топлив, полимеров, азотных удобрений и продукции малотоннажной химии.
В задачи проекта входит разработка новых типов катализаторов и технологий их производства; импортозамещение промышленных катализаторов нефтепереработки, нефте- и газохимии, азотной промышленности; оптимизация технологии производства известных типов катализаторов и разработка новых марок. Стоимость проекта оценивается в 3,5 миллиарда рублей. Примерная дата вывода объекта на проектную мощность – 2025 год.
Опытное производство катализаторов будет иметь формат центра коллективного пользования и разместится в двух новых корпусах на территории Института катализа СО РАН. После выхода опытного производства катализаторов на проектную мощность планируется изготовление не менее 20 партий образцов катализаторов в год.
В ближайшей перспективе и в среднесрочном периоде создаваемое Опытное производство катализаторов будет необходимо для выполнения трех проектов, получивших статус национальных в рамках реализации дорожной карты «Внедрение инновационных технологий и современных материалов в отраслях ТЭК» (Распоряжение Правительства РФ №1217-р от 03.08.2014):
В основе всех этих проектов лежат результаты ранее проведенных фундаментальных и прикладных исследований, характеризующихся новизной и значительным инновационным потенциалом.
Катализаторы гидропроцессов для глубокой переработки нефтяного сырья будут отличаться от известных импортных аналогов значительно меньшим содержанием дорогостоящих активных металлов и, соответственно, меньшей себестоимостью. Преимущество разрабатываемых катализаторов по сравнению с мировыми и отечественными аналогами будет заключаться в возможности снижения энергозатрат при производстве моторных топлив и увеличении выхода целевых продуктов – средних дистиллятов (бензина, керосина и дизельного топлива), соответствующих стандартам Евро 5 и 6, с улучшенными химмотологическими и низкотемпературными свойствами (арктическое и зимнее дизельное топливо).
В связи с модернизацией российских НПЗ потребность в катализаторах гидропроцесов существенно возрастает. Так, только к 2020 году потребность в этих катализаторах увеличится более чем в два раза (с 2,5 в 2014 году до 7,6 тысяч тонн в год). Уже к 2030 году в связи с увеличением мощностей гидропереработки вторичных нефтепродуктов потребности российских НПЗ в катализаторах гидропроцессов увеличатся в 4-5 раз и составят около десяти тысяч тонн в год. Создание на основе разработок ИК СО РАН отечественного производства катализаторов гидропроцессов на АО «Газпромнефть-ОНПЗ» сможет обеспечить не менее 70% потребностей российского рынка катализаторов.
Потенциальными потребителями разрабатываемых катализаторов гидропроцессов являются российские нефтеперерабатывающие компании, имеющие действующие в настоящее время установки гидроочистки дизельного топлива, гидроочистки и гидрокрекинга вакуумного газойля или заявившие о запланированном строительстве таких установок. Кроме ПАО «Газпром нефть», это ПАО «НК «Роснефть», ПАО «НК «Лукойл», ПАО «Татнефть», ПАО «Сургутнефтегаз», а также НПЗ Сербии, Беларуси, Сирии, Ирана, Венесуэлы.
На сегодняшний день потребности в катализаторах гидропроцессов обеспечиваются исключительно американскими и европейскими поставщиками, например, фирмами UOP, Albemarle и Axens. Вследствие санкций, вводимых США и ЕС, и возможного прекращения поставок импортных катализаторов установки гидропереработки могут полностью прекратить работу, что в целом по России приведет к сокращению объемов производства высококачественного моторного топлива (экологических стандартов Евро 4 и 5) более чем на 10 миллионов тонн в год.
Титанмагниевые катализаторы (ТМК) полимеризации олефинов применяются в производстве полимеров – полипропилена (ПП) и полиэтилена (ПЭ) различных марок. Прогнозируемое потребление ТМК в России к 2020 году увеличится в два раза по сравнению с 2017 и составит 150 тонн в год.
Разработки Института катализа характеризуются повышенным выходом полимеров (полиэтилена и полипропилена) на один килограмм катализатора – в 1,25 - 1,5 раза по сравнению с ныне существующими импортными катализаторами (отечественных аналогов нет). Кроме того, разработанные подходы к приготовлению ТМК позволяют производить новые модификации ТМК для производства полимеров современных марок.
В свою очередь, создание производства ТМК на базе отечественных разработок позволит обеспечить катализаторами полимеризации действующие производства полиэтилена и полипропилена, снизить риски при создании новых производств полимеров в России, сократить их 100%-ю зависимости от импорта ТМК.
Общая потребность в катализаторах ТМК для действующих производств ПП и ПЭ в РФ составляет более 70 тонн в год (стоимостью более 1,4 миллиарда рублей в год) и может быть удвоена с учетом строящихся и планируемых к вводу в эксплуатацию до 2020 года производств.
Гидроксид алюминия высокой чистоты предназначен для изготовления катализаторов риформинга бензиновых фракций и дегидрирования пропана в нефтеперерабатывающих и нефтегазохимических отраслях РФ.
Методы приготовления гидроксида алюминия, разработанные в ИК СО РАН, позволят получать шариковый носитель с требуемыми текстурными и прочностными характеристиками, обеспечивающий производство широкого ассортимента катализаторов процессов переработки углеводородов (риформинга в движущемся слое, дегидрирования пропана и др.) с лучшими каталитическими свойствами по сравнению с зарубежными аналогами.
Основными потребителями гидрооксида алюминия разной чистоты на территории РФ являются катализаторные производства. В настоящее время этот продукт в виде порошкообразного гидроксида алюминия высокой чистоты и готового шарикового (сферического) носителя катализатора закупается за рубежом в количестве до 490 тонн в год. Существует потенциал роста ежегодного спроса до 650 тонн к 2020 году, с учетом ввода в эксплуатацию новых установок риформинга с непрерывной регенерацией.
В настоящее время на территории РФ полностью отсутствует промышленное производство гидроксида алюминия высокой чистоты псевдобемитной структуры и шариковых носителей. Доля импорта данной продукции составляет 100%.
Для всей разрабатываемой в рамках национальных прооектов продукции предполагается использовать исключительно отечественное сырье. Так, для катализаторов гидропроцессов будут использоваться соединения алюминия и кремния, соли молибдена, кобальта, никеля и вольфрама, производимые отечественными компаниями.
Для ТМК будут использоваться соединения магния, титана отечественных производителей, например, ОАО «Соликамский магниевый завод» (г. Соликамск, Пермская область), ОАО «АВИСМА» (Березниковский титано-магниевый комбинат), ОАО «Реактив» (г. Санкт-Петербург).
Для производства гидроксида алюминия будет использоваться недорогой отечественный глинозем, в отличие от применяемого при алкоголятной технологии особо чистого алюминия, что существенно влияет на доступность исходного сырья и, в конечном счете, на себестоимость продукции.
Материал подготовили
А.М. Ершова, В.А. Яковлев
(Институт катализа СО РАН, Новосибирск)
Catalyst drives carbon-coupling chemistry without making CO2
Highly pure form of iron carbide may improve efficiency and lower cost of Fischer-Tropsch process
A simple synthesis method converts iron oxide to a highly pure form of iron carbide, yielding a catalyst for industrial carbon-coupling reactions that produces exceptionally low quantities of CO2, according to a study (Sci. Adv. 2018, DOI: 10.1126/sciadv.aau2947). The finding may improve efficiency and may also reduce the energy consumption and cost of operating Fischer-Tropsch (FT) reactors, which convert synthesis gas, or syngas (CO + H2), to liquid hydrocarbons such as diesel fuel. Iron- and cobalt-based catalysts both drive FT chemistry. Iron catalysts are less expensive, but they convert up to 30% of the CO in syngas to unwanted CO2, which lowers process efficiency and is costly to separate. So a team led by Emiel J. M. Hensen of Eindhoven University of Technology examined conventional iron FT catalysts and found that in addition to containing iron carbide, the desirable component, they also contain metallic iron and iron oxides, the CO2-forming culprits. The team evaluated preparation methods and found they could make highly pure iron carbide from low-cost iron oxide by fully reducing the starting material in hydrogen and then treating it with nitrogen-diluted syngas. Under industrial conditions, the new catalysts generated as little as 5% CO2 and remained stable for more than 150 hours.
Attached to MOFs, frustrated Lewis pair catalysts become recyclable
Tunable pairing opens new avenues for heterogeneous catalysis
Frustrated Lewis pairs (FLPs) stepped into the synthetic limelight just over a decade ago. Composed of a Lewis acid and a Lewis base whose blocked inclination to bond grants the molecules great powers, the main-group molecules were hailed for catalyzing hydrogenation reactions previously accomplished only with transition metals. The use of FLPs has been limited, however, by their lack of recyclability and their sensitivity to air and moisture.
Now, researchers led by Shengqian Ma at the University of South Florida have developed stable FLPs. Anchored inside a metal-organic framework (MOF), they efficiently catalyze hydrogenation and imine reduction reactions (Chem 2018, DOI: 10.1016/j.chempr.2018.08.018). With simple filtration, the catalyst can be recycled in the latter reaction at least seven times without loss of activity.
To construct the MOF-FLP pair, the team docked an amine Lewis base at the MOF’s open metal sites and then attached a boron Lewis acid (shown). The team then compared the heterogeneous MOF-FLP’s ability to catalyze reactions with that of a homogeneous catalyst. While the researchers observed comparable yields for the hydrogenation reaction, Ma says the MOF-FLP showed “interesting” steric and size selectivity in the imine reduction. The MOF-FLP matched the homogeneous catalyst’s high yield unless the substrate contained a buried imine, suggesting that the MOF environment restricts access to certain substrates. Substrates beyond a certain size were not reduced, likely because they can’t squeeze through the MOF’s pores, the authors say.

Catalysts that combined metal-organic frameworks and Lewis pairs were recycled
seven times or imine reduction reactions such as this one, without loss of activity.
“The beauty of MOFs is that they’re very tunable,” Ma says. Future MOF designs could easily introduce chirality or superhydrophobicity to expand the FLP catalysts’ reaction scope and durability, he says.
The University of Toronto’s Douglas W. Stephan, whose group pioneered the use of FLPs, calls the work a “remarkably clever and facile strategy.” The readily handled MOF-LPs avoid the air and moisture sensitivity of most homogeneous FLP catalysts, and their recyclability could also lower costs, Stephan says. The study, he adds, “foreshadows a vast potential for an array of new stable, recyclable, and selective FLP catalysts embedded in MOFs.”
Catalytic teamwork transforms alkenes selectively
Photocatalyst pairs with ene-reductase enzyme to obtain one enantiomer in reductions of mixtures

Double-bond-forming reactions commonly yield a mixture of alkene isomers with different geometric arrangements of functional groups. Subsequent transformations, such as reducing the double bond to a single bond, lead to mixtures that can be difficult to separate. John F. Hartwig of the University of California, Berkeley; Huimin Zhao of the University of Illinois, Urbana-Champaign; and their colleagues think strategic pairing can solve that problem and others. Catalyst pairs—both in biology and in industrial contexts—can carry out higher-yielding and more-selective reactions than can the individual catalysts in sequence. The team paired enzymes called ene-reductases with photocatalysts, which tend to be biocompatible because they often generate catalyst intermediates that are stable in water. The photocatalyst interconverts the two alkene isomers, and the awaiting enzyme selectively reduces one of the two isomers to a chiral product (Nature 2018, DOI: 10.1038/s41586-018-0413-7). The combination process also reduces single alkene isomers that would not react with the enzyme on its own (example shown). The team has filed a provisional patent application on the technology. A commentary accompanying the report notes that so far, the reaction requires substrates with aromatic groups, and the substrate concentrations are lower than industrial processes require.
Single atoms catalyze Suzuki reactions
Flow chemistry process takes place on carbon nitride surface

As effective as the palladium-catalyzed Suzuki reaction is for forming carbon-carbon bonds, researchers see room for improvement. Homogeneous palladium catalysts can be tough to separate from products, and fixing the problem by attaching catalysts to solid supports can lead to problematic metal leaching. Meanwhile, heterogeneous palladium nanoparticle catalysts on solid supports rarely catalyze the reaction efficiently. Now, Javier Pérez-Ramírez of ETH Zurich and colleagues suggest another route to Suzuki chemistry: single-atom catalysis (Nat. Nanotechnol. 2018, DOI: 10.1038/s41565-018-0167-2). Multiple labs have demonstrated catalysis of over 10 reactions by isolated atoms supported on a solid. This team anchored palladium atoms to nanometers-thick sheets of exfoliated graphitic carbon nitride, which contain metal-stabilizing nitrogen-rich sites. The catalyst performed a Suzuki reaction in flow for 13 hours with negligible metal leaching. Calculations from coauthor Núria López of the Institute of Chemical Research of Catalonia suggest that the palladium atoms continuously adapt their coordination within the same site on the carbon nitride as the reaction progresses, minimizing energy at each step. Experimentally verifying that result will be challenging, the authors say, but improvements to catalyst-monitoring spectroscopy techniques could help. The authors plan to test other reactions and metals with their system as well as launch a company to market the catalysts.
Nucleotide construction gets new chiral tool
Reagent developed through Scripps and BMS collaboration couples nucleosides with potential applications in antisense therapeutics
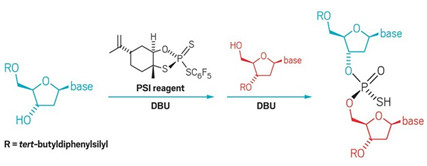
A
new phosphorus(V) reagent developed by researchers at BMS and Scripps
couples nucleosides diastereoselectively.
Antisense drugs are on the rise. The U.S. FDA has approved two antisense therapies to treat genetic diseases in the past two years with more in the pipeline. These compounds often consist of nucleoside chains connected by phosphorothioates—chiral linkages that make the agents more stable in the body yet exponentially increase their complexity. For example, the 18-mer Spinraza, which is a spinal muscular atrophy drug from Ionis and Biogen that costs $125,000 per dose, incorporates these linkages and is delivered as a mixture of more than 130,000 isomers theoretically.
Controlling the stereochemistry at phosphorus, though, could increase a drug candidate’s potency by reducing the number of less active isomers in these mixtures. To gain that control, chemists have mostly turned to phosphorus(III)-based chemistry. Now, scientists at Bristol-Myers Squibb and Scripps Research Institute California report a novel P(V)-based reagent that couples nucleosides with high diastereoselectivity (Science 2018, DOI: 10.1126/science.aau3369).
P(V) reagents are generally less reactive than P(III) but they let chemists skip cumbersome protection and deprotection steps required by P(III) chemistry, the authors say. Building on work by Wojciech J. Stec and colleagues, the team synthesized a P(V) reagent in one step from limonene oxide, which served as the chiral scaffold. The researchers point out that P(III)-based reagents are air and moisture sensitive and require specialized equipment to handle, while the new reagent is bench-stable, which they say could improve medicinal chemists ability to rapidly synthesize these molecules and discover new medicines.
The collaborators used their reagent to make a variety of dinucleotides in 61–97% yield. They also synthesized several oligonucleotides and cyclic dinucleotides, a class of compounds that has attracted attention as possible anticancer agents. The P(V) reagent produced a single diastereomer of a cyclic dinucleotide in five steps while the traditional P(III) approach produced four diastereomers in nine steps.
“It is exciting to see new developments in this challenging and important area of nucleotide chemistry,” says Jonathan Hall at ETH Zurich. “If it can be extended to pharmaceutically relevant oligonucleotide derivatives, it will represent a game-changer for oligonucleotide therapeutics.”
Chandra Vargeese, head of drug discovery at Wave Life Sciences, which develops stereopure nucleotides using a P(III)-based platform with industrial partners such as Takeda and Pfizer, says the approach is elegant and offers excellent diastereoselectivities on par with P(III) chemistry. The stepwise coupling yields, however, are on the low side compared to Wave’s platform and would need to be optimized for commercial production, she says. Another challenge for the P(V) reagent, Vargeese says, is that it doesn’t offer the same level of access as P(III) reagents do to structures with mixed phophorothioate and phosphodiester backbones that are important to the field.
The BMS-Scripps team says the P(V) reagent will soon be available for purchase through MilliporeSigma.
Chemical Engineering News
|
14-17
ноября 2018 г. III Всероссийская молодежная конференция “Проблемы и достижения химии кислород- и азотсодержащих биологически активных соединений” Уфа, Республика Башкортостан, Россия |
chemkonfbsu@gmail.com (347) 229-96-86 |
|
14-17
ноября 2018 г. Международная конференция “Современные инновации: химия и химическая технология ацетиленовых соединений. Нефтехимия. Катализ” Ташкент, Узбекистан |
confacetyleneuz@yandex.ru +9 9893 569 88 12 |
|
November
20-22, 2018 2nd International Conference on Functional Materials and Chemical Engineering (ICFMCE 2018) Abu Dhabi, United Arab Emirates |
http://www.icfmce.org |
|
December
6-8, 2018 International Conference on Materials for Energy Applications (ICME-18) Jaipur, India |
http://www.icme2018.subodhpgcollege.com |
|
December
9-13, 2018 6th International Conference on Metal-Organic Frameworks and Open Framework Compounds (MOF2018) Auckland, New Zealand |
http://mof2018.com |
|
December
10-12, 2018 International Conference on Phosphorus, Boron and Silicon 2018 Barcelona, Spain |
http://premc.org/conferences/pbsi-phosphorus-boron-silicon |
|
December
10-14, 2018 International Symposium on Catalysis and Fine Chemicals 2018 (C&FC 2018) Bangkok, Thailand |
http://www.cfc2018.org |
|
January 8-10, 2019 12th International Symposium on Advances in Electrochemical Science and Technology (iSAEST-12) Chennai, India |
http://krc.cecri.res.in/isaest/Home.html |
|
January
18-20, 2019 2nd International Joint Conference on Materials Science and Mechanical Engineering (CMSME 2019) Phuket, Thailand |
http://www.cmsme.net |
|
January
29, 2019 IYPT2019 Opening Ceremony: the International Year of the Periodic Table, the 150th Anniversary of the Periodic Table of Chemical Elements Paris, France |
http://www.iypt2019.org |
|
February
18-20, 2019 Hot-electron science and microscopic processes in plasmonics and catalysis – Faraday Discussion London, UK |
http://www.rsc.org/events/detail/27637/hot-electron-science-and-microscopic-processes-in-plasmonics-and-catalysis-faraday-discussion |
|
February
25-27, 2019 3rd International Conference on Catalysis and Chemical Engineering (Catalysis-2019) Houston TX, United States |
https://unitedscientificgroup.com/conferences/catalysis |
|
March
11-13, 2019 3rd International Congress on Catalysis and Chemical Science Singapore, Singapore |
https://catalysiscongress.com |
|
18-21
марта 2019 г. XXXV Всероссийский симпозиум молодых ученых по химической кинетике Московская обл., Россия |
http://www.chem.msu.ru/rus/ChemKin |
|
April
8-10, 2019 Applied Catalysis & Chemical Engineering Dubai, United Arab Emirates |
https://www.eigenscientificgroup.com/conference/catalysis-2019 |
|
April
9-12, 2019 26th Croatian Meeting of Chemists and Chemical Engineers (26HSKIKI) Šibenik, Croatia |
http://www.26hskiki.org/en |
|
May
5-8, 2019 4th Green & Sustainable Chemistry Conference Dresden, Germany |
https://www.elsevier.com/events/conferences/green-and-sustainable-chemistry-conference |
|
May
13-15, 2019 Conference “Frontiers in Catalysis and Chemical Engineering” (EURO CAT 2019) London, UK |
https://www.longdom.com/eurocat |
|
May
13-17, 2019 International Symposium on Green Chemistry (ISGC) La Rochelle, France |
https://www.isgc-symposium.com |
|
May
15-17, 2019 5th International Conference on Polygeneration (ICP 2019) Fukuoka, Japan |
http://therme.mech.kyushu-u.ac.jp/ICP2019/ICP2019.html (The website will be open in early May 2018) |
|
June
2-6, 2019 12th Natural Gas Conversion Symposium San Antonio, Texas, USA |
https://www.aiche.org/conferences/natural-gas-conversion-symposium/2019 |
|
June
2-6, 2019 14th International Symposium on Macrocyclic and Supramolecular Chemistry (ISMSC2019) Lecce, Italy |
https://ismsc2019.eu |
|
June
11-13, 2019 23rd Annual Green Chemistry & Engineering Conference and 9th International Conference on Green and Sustainable Chemistry Reston, VA (USA) |
http://gcande.org |
|
June
16-20, 2019 6th Nano Today Conference Lisbon, Portugal |
https://www.elsevier.com/events/conferences/nano-today-conference |
|
June
17-20, 2019 3rd International Conference on Applied Surface Science Pisa, Italy |
https://www.elsevier.com/events/conferences/international-conference-on-applied-surface-science |
|
June
24-28, 2019 5th EuChemS Inorganic Chemistry Conference Moscow, Russia |
http://www.igic.ras.ru/docs/news/tcirkulyar_evrohim_2019.pdf eicc5@ineos.ac.ru |
|
June
26-30, 2019 6th European Conference on Environmental Applications of Advanced Oxidation Processes (EAAOP-6) Portoroz-Portorose, Slovenia |
http://eaaop6.ki.si |
|
July
5-12, 2019 50th General Assembly & 47th IUPAC World Chemistry Congress Paris, France |
https://www.iupac2019.org |
|
July
8-11, 2019 14th International Conference on Materials Chemistry (MC14) Birmingham, UK |
http://www.rsc.org/events/detail/31760/14th-international-conference-on-materials-chemistry-mc14 |
|
July
15-19, 2019 Catalysis Fundamentals and Practice Summer School University of Liverpool, UK |
https://www.liverpool.ac.uk/chemistry/events/catalysis-summer-school-2019 |
|
July
17-19, 2019 Energy Security and Chemical Engineering Congress 2019 (SChE 2019) Penang, Malaysia |
http://esche.ump.edu.my/index.php/en |
|
July
26-28, 2019 Mendeleev 150: 4th International Conference on the Periodic Table endorsed by IUPAC Saint Petersburg, Russia |
http://mendeleev150.ifmo.ru |
|
August
4-8, 2019 36th International Conference of Solution Chemistry Xining, China |
http://icsc2019.csp.escience.cn/dct/page/1 |
|
August
18-23, 2019 European Congress on Catalysis (14th EuropaCat) Aachen, Germany |
www.europacat2019.eu |
|
September
2-6, 2019 1st International Conference on Noncovalent Interactions (ICNI) Lisbon, Portugal |
http://icni2019.eventos.chemistry.pt |
|
September
23-27, 2019 5th International Congress on Catalysis for Biorefineries (Catbior 2019) Turku / Åbo, Finland |
http://www.catbior2019.fi |
|
|
|
|
June
28 – July 3, 2020 The World Conference on Carbon (CARBON 2020) Kyoto, Japan |
http://www.tanso.org/carbon2020 |
|
July
5-9, 2020 48th World Polymer Congress (MACRO2020) Jeju Island, Korea |
http://www.macro2020.org |
|
August
16-21, 2020 12th Triennial Congress of the World Association of Theoretical and Computational Chemists (WATOC 2020) Vancouver, Canada |
http://watoc2020.ca |