
Российская академия наук
Отделение химии и наук о материалах
НАУЧНЫЙ СОВЕТ ПО КАТАЛИЗУ
Отчет о научно-организационной деятельности в 2014 году
Секретариат Научного совета по катализу ОХНМ РАН (НСК) предлагает Вашему вниманию
сводный отчет о деятельности Совета и научных исследованиях в области
катализа, выполненных научными коллективами под руководством членов
Научного совета по катализу.
Отчет состоит из трех разделов:
- Организационная деятельность,
- Отчеты по фундаментальным исследованиям, созданию новых каталитических систем и применению физических методов для их диагностики (в рамках
секции исследования механизмов каталитических реакций),
- Разработка и усовершенствование промышленных катализаторов и технологий (в рамках секции промышленного катализа).
Тексты отчетов, полученные от членов НСК и научно-исследовательских коллективов, практически не подвергнуты корректировке.
Организационная деятельность
В 2014 году в рамках научно-организационной деятельности Научного совета по катализу ОХНМ РАН (НСК) были выполнены следующие мероприятия.
1. Выпущены четыре ежеквартальных сборника «Каталитический бюллетень», содержащие оперативную информацию о важнейших
результатах фундаментальных и прикладных исследований в области катализа в России и за рубежом, материалы, посвященные деятельности
выдающихся отечественных и зарубежных исследователей в области катализа; дается перечень предстоящих конференций, краткие отчеты о проведенных конференциях, рабочих совещаниях и другие материалы.
2. Под эгидой Научного совета по катализу и при активном участии его членов организованы и проведены следующие конференции:
- V Всероссийская научная молодежная школа-конференция «Химия под знаком СИГМА», 12-18
мая 2014 г., Омск, Россия (организаторы: Институт проблем переработки углеводородов СО РАН, Институт катализа СО РАН)
- Научно-технологический симпозиум «Нефтепереработка: катализаторы и гидропроцессы»,
20-23 мая 2014 г., Санкт-Петербург, Россия (организаторы: Институт катализа СО РАН, Некоммерческое партнерство «Национальное каталитическое общество»)
- Заседание секции НТС ОАО «Газпром» «Комплексная переработка газа и газового конденсата». Катализаторы, адсорбенты и технологии их
использования в переработке природного газа. Проблемы и перспективы развития, 29 мая 2014 г., Новосибирск, Россия (организаторы: Институт катализа СО РАН, ОАО «Газпром»)
- XXI Международная конференция по химическим реакторам ХИМРЕАКТОР-21, 22-25 сентября 2014 г., Делфт, Нидерланды (организатор:
Институт катализа СО РАН)
- II Российский конгресс по катализу «РОСКАТАЛИЗ», 2-5 октября 2014 г., Самара, Россия (организаторы:
Институт катализа СО РАН, Самарский государственный технический университет, Московский государственный университет имени М.В. Ломоносова, Институт нефтехимического синтеза им. А.В. Топчиева РАН,
Институт органической химии им. Н.Д. Зелинского РАН, Институт проблем переработки углеводородов Сибирского отделения РАН)
Изданы материалы проведенных конференций.
Секретариат НСК ведет переписку и текущую работу с членами Научного совета по катализу ОХНМ РАН.
Продолжается сотрудничество с организациями Академий наук РФ и стран СНГ, Министерствами РФ, институтами разных ведомств и другими организациями России, дальнего и ближнего зарубежья по различным
вопросам научной, научно-организационной, учебно-преподавательской и общественной деятельности в области катализа.
ОСНОВНЫЕ РЕЗУЛЬТАТЫ 2014 г.
Фундаментальные исследования в области создания новых каталитических систем и применения физических методов для их диагностики
Катализ хиральными кислотами Льюиса
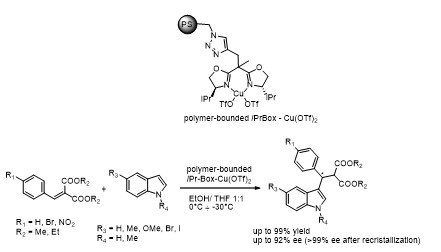
Осуществлен асимметрический вариант реакции Фриделя-Крафтса на примере реакции индола с арилиденмалонатами с помощью иммобилизованного на полимерную
матрицу катализатора состава iPr-Box/Cu(OTf)2; катализатор сохраняет активность в 5 циклах, продукты реакции получены с выходами до 99% и с энантиомерными избытками до 92% ее,
а после перекристаллизации энантиомерная чистота была увеличена до 99% ее.
академик И.П. Белецкая
Институт физической химии и электрохимии
им. А.Н. Фрумкина РАН, г. Москва
Исследование формирования Fe- и Co-содержащих
наноразмерных катализаторов Фишера-Тропша
Определены оптимальные условия формирования Fe- и Co-содержащих наноразмерных катализаторов Фишера-Тропша, полученных in situ в реакционной среде в условиях трехфазного процесса в сларри-реакторе.
Варьировались такие параметры синтеза как природа дисперсионной среды, природа дисперсной фазы, природа и концентрация прекурсора железа, а также природа и концентрация ПАВ. Выявлены зависимости
размера частиц эмульсий от природы и концентрации оксидных промоторов. Установлены оптимальные условия восстановительного термолиза эмульсии in situ в сларри-реакторе для получения
стабильных наноразмерных Fe- и Co-содержащих суспензий.
академик С.Н. Хаджиев, к.х.н. М.В. Куликова
Институт нефтехимического синтеза
им. А.В. Топчиева РАН, г. Москва
Приготовление силикоалюмофосфатных катализаторов для синтеза низших олефинов из метанола
Получены силикоалюмофосфатные катализаторы для синтеза низших олефинов из метанола с повышенной стабильностью
работы во времени. Эта цель была достигнута за счет модифицирования внешней поверхности
катализаторов либо покрытием поверхности кристаллов SAPO аморфным оксидом кремния (до 2 масс. % по кремнию), либо методом рекристаллизации, создающим на поверхности силикоалюмофосфата слой мезопористой фазы. Данный
подход к модифицированию катализаторов позволил покрыть/дезактивировать поверхностные кислотные центры, ответственные за олигомеризацию низших олефинов. Опыты по конверсии метанола в
низшие олефины показали, что модифицирование позволяет увеличить время стабильной работы катализатора в 2 раза, а также повысить селективность по олефинам С2-С4
за счет снижения вклада продуктов С5+.
академик С.Н. Хаджиев, д.х.н. И.И. Иванова
Институт нефтехимического синтеза
им. А.В. Топчиева РАН, г. Москва
Мембранно-каталитический генератор синтез-газа и водорода, совмещённый с топливными элементами
Для малогабаритных электростанций нового поколения в ИНХС РАН (руководитель работы д.х.н. М.В. Цодиков) совместно с ИСМАН РАН (к.т.н. В.И. Уваров) разработаны пористые мембранно-каталитические
конвертеры, проявляющие повышенную активность в паровом и углекислотном риформинге метана или продуктов ферментации биомассы (в том числе без их разделения на индивидуальные компоненты). Конверторы
апробированы для работы с топливными элементами любого типа (Институт высокотемпературной электрохимии УРО РАН, руководитель д.х.н. Зайков Ю.П.). Пористые мембранно-каталитические системы, внутренние каналы
которых модифицированы наноразмерными катализаторами, позволяют переработать органические субстраты в синтез-газ и водород при малом времени контакта – 0,05-0,1 с, благодаря чему достигается
рекордная производительность по синтез-газу (на уровне 85000 л/ч дм3мембр). На модельной лабораторной установке, где конвертор интегрирован с высокотемпературным твердооксидным ТЭ, были достигнуты высокие
показатели по выработке электроэнергии (I – 300 мА/см2, W – 150 мВт/см2).
Один конвертер с габаритами 14×1,5 см способен обеспечить первичным топливом (синтез-газ) до 10 ТЭ мощностью до 100 Вт. В развитие этого направления разработаны оригинальные гибридные
конвертеры, способные обеспечить очищенным и ультрачистым водородом ТЭ, работающие при средних (120-150°C) и низких (20-30°C) температурах. Для среднетемпературных ТЭ содержание СО в водороде не
должно превышать 2%. В этом случае во внутреннем объеме гибридного конвертера размещают гранулированный катализатор парового риформинга СО. Для низкотемпературных ТЭ разработан и испытан оригинальный
конвертер, который во внутреннем объеме содержит мембрану в виде трубки, изготовленную из палладий-содержащего сплава. В этом конвертере наряду с синтез-газом состава Н2/СО
– 2,7 из реакционной зоны выделяется до 52% ультрачистого водорода.
д.х.н. М.В. Цодиков, к.х.н. А.С. Федотов, Д.О. Антонов,
к.т.н. В.И. Уваров, д.х.н. В.Н. Корчак, академик И.И. Моисеев
Институт
нефтехимического синтеза им. А.В. Топчиева РАН, г. Москва
Институт структурной макрокинетики и проблем материаловедения РАН, г. Черноголовка
Институт общей и неорганической химии
им. Н.С. Курнакова РАН, г. Москва
Восстановительная деструкция липидов рапсового масла в узкие фракции
углеводородов С3 и С18
Одним из приоритетных направлений современной химической энергетики является разработка высокоэффективных процессов переработки
возобновляемого сырья в ценные мономеры и топливные компоненты.
В 2014 г. совместно ИНХС РАН и ИОНХ РАН разработан катализатор селективного превращения рапсового масла в узкие алкан-олефиновые
фракции. На основе гетерометаллического Pt-Sn-содержащего комплекса разработан катализатор, содержащий биметаллические активные
компоненты на поверхности γ-Al2O3. Установлено, что при соотношении активных компонентов Sn/Pt = 5
триглицериды жирных кислот рапсового масла селективно превращаются в узкие фракции углеводородов С3 и С18.
Показано, что глицериновый фрагмент более чем на 30% превращается в пропан-пропиленовую фракцию, а углеводородные фрагменты жирных
кислот более чем на 90% – в алкан-олефиновую фракцию С18. Кислород триглицерида жирных кислот селективно восстанавливается в
воду, в результате чего выход «тупиковых» С1 и С2 в этом процессе не превышает 0,7%. В составе фракции
углеводородов С18 содержится до 22% ценных линейных олефинов. Изучение структуры активных компонентов методом ПЭМ ВР
показало, что при соотношении активных компонентов Sn/Pt = 5 размеры активных металлсодержащих компонентов на поверхности носителя
≤ 1 нм. При столь малом размере активных компонентов катализатора взаимодействие ТГЖК с активными центрами протекает лишь
по кислородным атомам молекулы липида, приводя к селективной деоксигенации и практически не затрагивая углеводородные фрагменты
молекулы. Таким образом, полученный результат позволяет минимизировать потери углеводородной компоненты исходной биомассы при
ее переработке в ценные компоненты топлив и мономеры.
д.х.н. М.В. Цодиков, к.х.н. А.В. Чистяков, П.А. Жарова,
к.х.н. С.С. Шаповалов, д.х.н. А.А. Пасынский, к.х.н. В.Ю. Мурзин,
чл.-корр. РАН А.Е. Гехман, академик И.И. Моисеев
Институт нефтехимического синтеза им. А.В. Топчиева РАН,
Институт общей и неорганической химии им. Н.С. Курнакова РАН, г. Москва
Создание высокочувствительной установки, обеспечивающей in situ контроль топохимических превращений при синтезе катализаторов
Разработана и введена в эксплуатацию высокочувствительная установка, обеспечивающая in situ контроль топохимических превращений при синтезе катализаторов, содержащих наночастицы
ферромагнитных металлов. Установка выполнена на базе вибрационного магнитометра и обеспечивает рабочие температуры в зоне реакции от 300 до 870 К при пропускании сквозь реакционную зону газов (смеси
газов) со скоростью до 150 см3/мин. Приготовлены высокоактивные катализаторы синтеза Фишера-Тропша на основе Fe-содержащих композитных материалов. Разработан аппаратурный комплекс, позволяющий
проводить исследования механизмов синтеза цеолитных катализаторов с помощью ЯМР под давлением и в агрессивных средах, установлены общие
принципы и конкретные методические подходы метода ЯМР ВМУ in situ для исследования гидротермального синтеза и пост-синтетического модифицирования
цеолитных и композитных катализаторов на их основе. Разработанный подход лег в основу проекта РНФ, направленного на создание научных основ рационального дизайна катализаторов важнейших процессов
нефтегазового комплекса, включающих крекинг, алкилирование, олигомеризацию, дегидратацию и конденсацию углеводородов и спиртов.
академик В.В. Лунин, Н.Н. Кузнецова
Московский государственный университет имени М.В. Ломоносова,
Химический факультет, г. Москва
Разработка способов получения высокоэффективных катализаторов восстановительных и окислительных реакций на основе биотемплатов
В рамках соглашения с РНФ 14-33-00018 методом пиролиза древесных опилок, пропитанных нитратом палладия, получены катализаторы Pd/C,
содержащие на поверхности, по данным рентгеновской фотоэлектронной спектроскопии, только восстановленные наноразмерные частицы палладия
(2-4 нм), нанесенные на активированный уголь, сохраняющий элементы пористой структуры древесины. Применение дополнительной обработки
опилок перед пиролизом позволило увеличить удельную поверхность получаемых систем с 7 м2/г
(без обработки) до 230 м2/г (обработка водой в гидротермальных условиях). Полученные каталитические системы проявили высокую активность в реакции
гидродехлорирования хлорбензола в паровой фазе в интервале температур 150-350°C.
В рамках проекта РФФИ 13-03-00613 с использованием цетилтриметиламмонийбромида или древесных опилок в качестве темплата получены системы CuO-CeZrO2,
проявляющие заметную активность в окислении СО в широком температурном диапазоне (100-450°C) и имеющие развитую пористую структуру, обусловленную темплатом.
Методами рентгенофазового анализа и рентгеновской фотоэлектронной спектроскопии показано, что оксид меди присутствует в виде отдельной фазы, а другие компоненты образуют сложный оксид.
академик В.В. Лунин, д.х.н. Е.С. Локтева,
к.х.н. Е.В. Голубина, К.И. Маслаков
Московский государственный университет имени М.В. Ломоносова,
Химический факультет, г. Москва
Новый метод приготовления катализаторов синтеза Фишера-Тропша. Гидроксикарбонилирование стирола в присутствии комплексных катализаторов
Предложен новый метод приготовления катализаторов синтеза Фишера-Тропша, заключающийся в нанесении металлического кобальта на носитель в
момент восстановления из солей раствором NaBH4.
Гидроксикарбонилированием стирола получены фенилпропионовые кислоты с выходом 97,5% при соотношении Pd : стирол = 1 : 200 и температуре 100°C.
Селективность по целевой 2-фенилпро-пионовой кислоте 98,8%. Впервые обнаружена высокая каталитическая активность комплексов HRh(CO)(PPh3)3,
RhCl(CO)(PPh3)2 в реакции восстановительного карбонилирования иодобензола.
чл.-корр. РАН А.Л. Лапидус
Институт органической химии им. Н.Д. Зелинского, г. Москва
Каталитическая активность систем на основе нанесённых калиевых солей карбонилгидридов переходных металлов в дейтероводородном обмене углеводородов
Установлено, что при нанесении K2[Ru4(CO)13], K2[Os3(CO)11], K2[Fe2(CO)8]
и KRe(CO5) на уголь «Сибунит» с последующим термическим разложением нанесённого карбонилметаллата в токе водорода или аргона образуются системы, способные катализировать реакции перераспределения изотопов
водорода в метане, этилене и ацетилене, а также дейтероводородный обмен между метаном и D2. В случае этилена и ацетилена изотопный обмен на наиболее активных
катализаторах протекает с высокой скоростью уже при комнатной температуре, а в случае метана – начиная со 150°C. На основании данных ГР-спектров сделан вывод, что активность
вышеуказанных железных катализаторов в H/D обмене обусловлена железосодержащими аморфными сплавами не установленной пока природы, образующимися при термическом разложении нанесённого K2[Fe2(CO)8].
α-Железо в ходе распада K2[Fe2(CO)8] в токе H2 или Ar практически не образуется. Таким образом, впервые продемонстрирована возможность использования систем на основе
нанесённых карбонилметаллатов калия для активации C–H-связей
углеводородов.
д.х.н. В.Б. Шур, С.М. Юнусов, З. Руммель, М. Херман, Е.С. Калюжная
Институт элементоорганических соединений им. А.Н. Несмеянова РАН, г. Москва
Институт модификации поверхности им. Лейбница,
Лейпциг, Германия
Моделирование структуры, свойств ферментов и катализируемых ими химических превращений
Проведена серия работ по моделированию структуры, свойств ферментов и катализируемых ими химических превращений многоуровневыми методами
квантовой механики/молекулярной механики (КМ/ММ). Характеризован полный каталитический цикл гидролиза пенициллина G ферментом пенициллинацилазой;
предложены оригинальные способы ингибирования матриксных металлопротеиназ in situ; объяснена роль канцерогенных точечных мутаций в белке Ras при гидролизе гуанозинтрифосфата
белковым комплексом Ras-GAP.
чл.-корр. РАН С.Д. Варфоломеев, Немухин А.В., Григоренко Б.Л.
Институт биохимической физики им. Н.М. Эмануэля РАН, г. Москва
Биокатализаторы для получения органических кислот из возобновляемого углеродосодержащего сырья
В рамках проекта «Биокаталитическое получение органических кислот из непищевого сырья как мономеров для органического синтеза
биопластиков» разработана серия оригинальных высокоактивных биокатализаторов (7 шт.) в виде иммобилизованных в криогель поливинилового спирта клеток
микроорганизмов для получения органических кислот (янтарной, яблочной, фумаровой, аспарагиновой) из разнообразного возобновляемого углеродосодержащего сырья, способных функционировать длительное время
(до 2000 ч) в различных исходных по характеристикам средах с максимальным выходом целевого продукта до 90% от теоретически возможного уровня. Впервые установлена эффективность использования в
качестве сырья гидролизатов целлюлозосодержащей биомассы, биомассы фототрофных микроорганизмов и макроводорослей и глицерина (отхода производства биодизеля) для получения органических кислот с
использованием разработанных биокатализаторов. По широте спектра образцов возобновляемого сырья, конвертируемых в фумаровую и янтарную кислоты, разработанные биокатализаторы мировых
аналогов не имеют.
чл.-корр. РАН С.Д. Варфоломеев
Институт биохимической физики им. Н.М. Эмануэля РАН,
Кафедра химической энзимологии Химического факультета МГУ им. М.В. Ломоносова, г. Москва
Разработка метода синтеза легкого аналога биодизеля путем биокаталитической переработки растительного сырья
В рамках Программы фундаментальных исследований Президиума РАН «Энергетические аспекты глубокой переработки ископаемого и возобновляемого углеродсодержащего сырья» разработан метод
синтеза легкого аналога биодизеля, состоящего из этиловых эфиров уксусной, пропионовой, масляной кислот, получаемых в результате биокаталитической переработки углеводистых отходов растительного
происхождения. Проведены испытания эфиров в составе топливных композиций, зарегистрирован значительный октанповышающий эффект. Исследуется возможность применения
сверхкритической флюидной технологии к синтезу биодизеля и его легкого аналога на основе низших жирных кислот, продуцируемых при биокаталитической переработке растительного сырья. Имеющийся задел
позволяет прогнозировать большую экономичность указанных процессов за счет сокращения их длительности, снижения расхода реагентов, отказа от использования катализатора.
чл.-корр. РАН С.Д. Варфоломеев, В.Б. Вольева, И.С. Белостоцкая, Н.Л. Комиссарова, А.В. Малкова
Институт биохимической физики им. Н.М. Эмануэля РАН, г. Москва
Исследование кинетики биомиметического окисления алканов на основе порфириновых комплексов металлов и бензальдегида
Проведено исследование кинетики биомиметического окисления алканов в системах на основе порфириновых комплексов металлов и бензальдегида.
Установлено, что в процессе окисления алканов, катализируемом порфиринами железа и марганца, в исследованных системах параллельно образуются спирт, кетон и гидропероксид. Обнаружено
влияние лигандного окружения металла на соотношение молекулярного и радикального маршрутов окисления алканов в этих биомиметических системах. Полученные результаты являются важным шагом на пути
создания теоретической базы для разработки методов селективного окисления алканов.
чл.-корр. РАН С.Д. Варфоломеев, Е.И. Карасевич
Институт биохимической физики им. Н.М. Эмануэля РАН, г. Москва
Установление механизма катализа комплексами {LiSt•DMF(HMPA)} в селективном окислении этилбензола молекулярным кислородом
Методом ГЖХ (с привлечением компьютерных программ обработки экспериментальных данных Mathcad and Graph2Digit) установлен механизм катализа комплексами
{LiSt•DMF(HMPA)} в селективном окислении этилбензола молекулярным кислородом в α-фенилэтилгидропероксид (ФЭГ) (селективность SФЭГ = 90-93%, конверсия
в ФЭГ C = 20-23%), являющийся полупродуктом многотоннажного производства мономеров: пропиленоксида и стирола. Рост параметров SФЭГ
и C при катализе комплексами {LiSt•DMF(HMPA)} по сравнению с катализом LiSt и некатализированным окислением этилбензола связан с изменением механизма образования побочных
продуктов – ацетофенона (АФ) и метилфенилкарбинола (МФК): последовательное (из ФЭГ) → параллельное ФЭГ, и торможением
гетеролиза ФЭГ. Впервые с помощью метода АСМ продемонстрирована возможность формирования супрамолекулярных наноструктур на основе
комплексов Ni(acac)2•ГМФА•PhOH(«А») за счёт межмолекулярных H-связей, что позволяет объяснить стабильность катализатора («А») в
процессе селективного окисления этилбензола в ФЭГ, катализируемом «А».
чл.-корр. РАН С.Д. Варфоломеев, Л.И. Матиенко, Л.А. Мосолова
Институт биохимической физики им. Н.М. Эмануэля РАН, г. Москва
Катализаторы глубокой гидроочистки тяжелого вакуумного газойля
Продолжаются исследования и разработки новых катализаторов глубокой гидроочистки нефтяных фракций и остатков. Разработаны составы и способы синтеза
Co(Ni)Mo и NiW катализаторов на основе широкопористого Al2O3, полученного с использованием мезоструктурированного гидроксида
алюминия. Катализаторы позволяют провести глубокую гидроочистку тяжелого вакуумного газойля до остаточного содержания серы менее 500 ppm при 360°C,
давлении водорода 5 МПа, объемной скорости подачи сырья 1.0 ч–1 и Н2/сырье 800 нл/л.
к.х.н. П.А. Никульшин, П.П. Минаев, Ал.А. Пимерзин, к.х.н. А.В. Можаев,
д.х.н. А.А. Пимерзин
Самарский государственный технический университет, г. Самара
Изучение закономерностей «состав-структура-свойство» в катализе сульфидами переходных металлов
Продолжаются исследования способов направленного формирования активной фазы сульфидных катализаторов гидроочистки. Исследовано влияние состава
NiWS катализаторов, природы и текстурных характеристик носителя на каталитические свойства в реакциях гидрообессеривания, гидрирования и
гидродеазотирования. Удельные активности катализаторов возрастают с увеличением содержания Ni в составе частиц NiWS фазы и их дисперсности. Показано, что частота оборотов в изученных реакциях
растет с уменьшением средней длины частиц активной фазы и степени промотирования ребер кристаллитов WS2.
к.х.н. П.А. Никульшин, П.П. Минаев, к.х.н. А.В. Можаев, М.С. Куликова, д.х.н. А.А. Пимерзин
Самарский государственный технический университет, г. Самара
Катализаторы для совместной гидроочистки нефтяного и растительного (возобновляемого) органического сырья
Ведутся исследования и разработки новых сульфидных катализаторов совместной гидроочистки нефтяных фракций и растительного (возобновляемого) сырья
с целью возможного расширения сырьевой базы нефтепереработки. В зависимости от природы носителя частота оборотов для СоМо центров катализаторов снижается
в ряду: SiO2 > Al2O3 > ZrO2 ≈ TiO2. Дальнейшее совершенствование катализаторов гидродеоксигенации может развиваться
путем снижения длины частиц активной фазы, увеличивая содержание активных центров и оптимизируя степень декорирования промотором кристаллитов MoS2.
к.х.н. П.А. Никульшин, В.А. Сальников, к.х.н. А.В. Можаев, А.Н. Варакин, д.х.н. А.А. Пимерзин
Самарский государственный технический университет, г. Самара
Создание эффективных магнитноотделяемых катализаторов на основе коммерчески доступных лигандов
Магнитные наночастицы могут использоваться в каталитических системах для магнитного отделения катализаторов от реакционной среды. Один из
известных методов синтеза монодисперсных частиц оксида железа или феррита кобальта, которые стабильны на воздухе, это высокотемпературное разложение кислородсодержащих соединений железа и
кобальта, таких как ацетилацетонаты, ацетаты или олеаты в растворах, содержащих ПАВ. За отчетный период разработаны способы формирования магнитных наночасти в полимерных матрицах, а именно в дендримерах и
сверхсшитом полистироле. Узкое распределение магнитных наночастиц по размерам обеспечивается благодаря разделению по температурам зародышеобразования и роста наночастиц. В зависимости от реакционных
условий можно варьировать размер, форму и кристаллическую структуру таких наночастиц, а следовательно, их магнитные свойства. Разработаны методики синтеза наночастиц каталитически активных металлов, таких
как рутений, платина, палладий, иммобилизованных на магнитные частицы. Магнитноотделяемые катализаторы протестированы в реакциях селективного гидрирования и окисления.
Разработаны новые подходы для получения материалов, содержащих одновременно магнитные и каталитические частицы. При этом обеспечивается высокая
магнитная восприимчивость таких материалов. С другой стороны, это не сказывается отрицательно на свойствах каталитических частиц, а наоборот, усиливает каталитическую активность и селективность.
Магнитные свойства разрабатываемых каталитических систем позволят быстрое и количественное отделение катализаторов от реакционных растворов, обеспечивая многократное использование катализаторов, приводящее к
удешевлению процессов и соответствующих продуктов, существенным энергосбережениям благодаря исключению стадии фильтрации и улучшению качества целевых продуктов.
д.х.н., проф. Э.М. Сульман
Тверской государственный технический университет, Институт нано- и биотехнологий, г. Тверь
Синтез пиридинов на цеолитных катализаторах
С целью разработки селективных гетерогенно-каталитических способов получения пиридина и его метилпроизводных – важных промежуточных соединений для синтеза фармацевтических препаратов,
гербицидов, ингибиторов коррозии металлов, ускорителей вулканизации каучука, традиционных лигандов при химической сборке координационных соединений – исследован в проточном реакторе синтез пиридинов с
участием этанола, формальдегида и аммиака в присутствии цеолитных катализаторов различного структурного типа: H-Beta (структурный тип
BEA), H-ZSM-12 (MTW), H-ZSM-5 (MFI). Установлено, что наиболее активным в синтезе пиридинов является цеолит H-Beta, на котором конверсия этанола достигает 60-70% (300-400°C,
2 ч–1). Традиционно используемый для синтеза пиридинов пентасил H-ZSM-5 в изученных условиях проявляет
меньшую активность (конверсия спирта 30-40%). Обнаружено, что основными продуктами реакции на цеолитах H-Beta и H-ZSM-5 являются
пиридин и метилпиридины, на цеолите H-ZSM-12 дополнительно образуется до 20% диметилпиридинов. Селективность образования пиридина максимальна (49%) при 200°C, 2 ч–1.
Показано, что повышение температуры, уменьшение объемной скорости и концентрации формальдегида в сырьевой смеси приводит к повышению в составе продуктов реакции количества метилпиридинов и «тяжёлых»
соединений.
чл.-корр.
РАН У.М. Джемилев, д.х.н. Н.Г. Григорьева, д.х.н. Б.И. Кутепов
Институт нефтехимии и катализа РАН, г. Уфа
Новые аминометилирующие реагенты в каталитическом синтезе макрогетероциклов
В развитие проводимых исследований по синтезу новых N,S-макрогетероциклов, перспективных в качестве биологически активных соединений, сорбентов
благородных и редких металлов, разработан эффективный метод синтеза 3-арил-1,5,3-дитиазацикланов и 6-арил-1,11-диокса-4,8-дитиа-6-азациклотридеканов
циклоаминометилированием α,ω-дитиолов (бутан-1,4-, пентан-1,5-, гексан-1,6-дитиолы, 3,6-диоксаоктан-1,8-дитиол) с помощью N,N-бис(метоксиметил)-N-ариламинов с участием Sm- и
Сu-содержащих катализаторов. Установлено, что при взаимодействии N,N-бис(метоксиметил)-N-фениламина с эквимольным количеством бутан-1,4-дитиола в условиях (~ 20°C,
EtOAc, 6 ч) с использованием 5 мол % катализатора Sm(NO3)3/γ-Al2O3 образуется 3-фенил-1,5,3-дитиазонан с выходом ~90%. В
разработанных условиях [5 мол % Sm(NO3)3/γ-Al2O3, 20°C, 6 ч] в реакцию циклоаминометилирования с бутан-1,4-дитиолом были
вовлечены N,N-бис(метоксиметил)-N-арил(м-метилфенил-, м-метоксифенил-, п-хлорфенил-, м-бромфенил- и п-бромфенил)амины
с получением соответствующих 3-арил-1,5,3-дитиазонанов с выходами 85-90%. С участием гетерогенного катализатора Sm(NO3)3/γ-Al2O3 осуществлен
селективный синтез 3-арил-1,5,3-дитиазеканов и 3-арил-1,5,3-дитиазациклоундеканов с выходами 75-80%. Катализируемое CuCl циклоаминометилирование α,ω-алкандитиолов и 3,6-диокса-1,8-октандитиола с помощью
N,N-бис(метоксиметил)-N-ариламинов проходит по типу [1+1] циклоконденсации и позволяет осуществить одностадийный синтез перспективных N-арилзамещенных
1,5,3-дитиазамакрогетероциклов различной структуры с высокими выходами.
чл.-корр. РАН У.М. Джемилев, к.х.н. Н.Н. Махмудиярова, асп. Л.В. Мударисова, д.х.н., проф. А.Г. Ибрагимов
Институт нефтехимии и катализа РАН, г. Уфа
Эффективный малостадийный синтез алмазоподобных углеводородов с использованием неорганических ионных жидкостей в качестве суперкислотных катализаторов
Разработаны новые, малостадийные методы синтеза алмазоподобных углеводородов – диамантана и триамантана. Сокращение числа стадий синтеза достигнуто
благодаря использованию новых С14H20 и С18H24 углеводородов – прекурсоров, представляющих легкодоступный димер норборнадиена
экзо-экзо-гексацикло[9.2.1.02,10.03,8.04,6.05,9]тетрадецена-12 и его [4+2]-аддукта с бутадиеном.
Установлено, что эффективными катализаторами скелетной изомеризации указанных углеводородов в диамантан и триамантан являются неорганические ионные жидкости [AlCl3]n·[R3N]m·HCl,
которые позволяют провести процесс в мягких условиях (40-50°C, 8 ч) и с высоким выходом, 82 и 80%, соответственно.
Доступность исходных мономеров, высокие выходы целевых каркасных соединений, простота осуществления реакций делают эти методы исключительно перспективными для синтеза уникальных мономеров для специальных
полимерных материалов, прекурсоров для получения противовирусных лекарственных препаратов.
д.х.н., проф. Р.И. Хуснутдинов, к.х.н. Р.И. Аминов, чл.-корр.РАН У.М. Джемилев
Институт нефтехимии и катализа РАН, г. Уфа
Новые реакции для направленной функционализации углеродных кластеров с участием комплексов титана
В развитие проводимых в лаборатории фундаментальных исследований по разработке эффективных методов введения в молекулы углеродных кластеров функциональных групп, базирующихся на выдвинутой нами ранее
идее о возможности использования фуллеренов в качестве олефиновой компоненты в реакции Кулинковича, впервые показано, что нитрилы вступают в реакцию с С60-фуллереном
в присутствии небольшого избытка EtMgBr и катализатора Ti(Oi-Pr)4 (5 мол.%) c селективным образованием ранее не описанных фуллеротетрагидропиридинов с выходами 15-40%.
Замена нитрилов на алкилиденцианоацетаты в данной реакции позволяет направить указанную реакцию в сторону формирования ранее труднодоступных индивидуальных фуллероаминоциклопропанов, что создает
перспективы для разработки на основе упомянутых выше производных фуллеренов промышленно важных материалов и веществ с полезными свойствами.
чл.-корр. РАН У.М. Джемилев, асп. З.Р. Шакирова, к.х.н. А.Р. Туктаров
Институт нефтехимии и катализа РАН, г. Уфа
Бестемплатный синтез мезопористых каталитически активных алюмосиликатов
Разработан бестемплатный способ золь-гель синтеза мезопористых каталитически активных алюмосиликатов с использованием тетраэтилортосиликата или
его олигомерных эфиров и водно-спиртового раствора нитрата алюминия. Способ позволяет синтезировать мезопористые материалы с весьма узким распределением пор, однородной структурой и высокой кислотностью
поверхности. Показано, что синтезированные алюмосиликаты являются эффективными катализаторами кислотно-основного типа, перспективными
для применения в таких промышленно значимых реакциях, как олигомеризация различных олефинов и алкилирование фенолов олефинами.
чл.-корр. РАН У.М. Джемилев, д.х.н. Н.Г. Григорьева, асп. М.Р. Аглиуллин, д.х.н. Б.И. Кутепов
Институт нефтехимии и катализа РАН, г. Уфа
Нанокристаллические оксидные вольфрамовые бронзы, полученные электролизом расплавов, в каталитических процессах окислительного обессеривания и облагораживания нефтяных фракций
Синтезированы новые гибридные органо-неорганические материалы – нанокристаллические оксидные вольфрамовые бронзы на углеродных поверхностях в виде гексагональных и тетрагональных кристаллических
структур. Полученные материалы проявляют на порядок большую каталазную активность по сравнению с порошковым материалом.
Результаты работы опубликованы в следующей статье: Петров Л.А., Шишмаков А.Б., Вакарин С.В., Семерикова О.Л., Меляева А.А., Микушина Ю.В., Зайков Ю.П., Чупахин О.Н. Поведение наноразмерных
оксидных вольфрамовых бронз, полученных высокотемпературным электролизом в модельных процессах обессеривания нефтепродуктов // Журнал неорганической химии. 2014. Т. 59. № 1. С. 72–75.
академик О.Н. Чупахин, д.х.н. Л.А. Петров, к.т.н. А.В. Шишмаков
Институт органического синтеза им. И.Я. Постовского УрОРАН, г. Екатеринбург
Новые катализаторы асимметрического синтеза
С использованием методологии прямого нуклеофильного замещения водорода в азааренах (SNH реакции) получены новые продукты
некатализируемого переходными металлами С-С кросс-сочетания азинов с хиральными производными ферроценов. Синтезированные оптически активные ферроценилазины в виде комплексов с палладием показали
высокую эффективность как катализаторы асимметрического синтеза в аллильном алкилировании олефинов по Тсуджи-Тросту (ее 97-99%).
академик О.Н. Чупахин, к.х.н. И.А. Утепова, к.х.н. А.А. Мусихина, асп. П.О. Серебренникова
Институт органического синтеза им. И.Я. Постовского УрОРАН
Уральский федеральный университет имени первого Президента России Б.Н. Ельцина, г. Екатеринбург
Синтез нанесенных платиновых катализаторов с использованием слоистых двойных гидроксидов различного катионного состава и их исследование в реакции дегидрирования пропана
Проведено варьирование содержания двух- и трехвалентного модифицирующего компонента (Zn, Ga) в составе слоистых двойных гидроксидов (предшественников полиметаллического оксидного носителя некислотного
типа для платиновых катализаторов). Установлено, что введение Zn и Ga в состав носителя обеспечивает более высокие значения конверсии пропана (при селективности 99%) при
использовании катализаторов 0.3%Pt/Mg(Zn)AlOx и 0.3%Pt/MgAl(Ga)Ox по сравнению с платиной, нанесенной на алюмомагниевый носитель. Для получения оптимальных
значений каталитических характеристик доля модификатора в составе двух- или трехвалентных катионов составляет 0.1-0.3. Результаты важны для нефтехимической промышленности в области производства олефинов.
чл.-корр. РАН В.А. Лихолобов, к.х.н. А.В. Лавренов
Институт проблем переработки углеводородов СО РАН, г. Омск
Исследование палладиевых катализаторов на различных углеродных носителях в реакции селективного гидрирования фурфурола до фурфурилового спирта
Установлено влияние типа углеродного носителя на формирование, состояние и каталитические свойства металлических центров катализаторов аквафазного гидрирования фурфурола. Было показано, что природа
носителя (наноглобулярный углерод (НГУ) и углеродные нанотрубки (УНТ)) влияет на размер частиц и зарядовое состояние нанесенного палладия и его активность в реакции гидрирования фурфурола.
Катализатор Pd/НГУ проявил наибольшую селективность в образовании фурфурилового спирта (99%) в мягких условиях реакции (50°C, 5 атм). В то же время образец Pd/УНТ
оказался неактивным в тех же условиях, однако способствовал восстановлению фуранового кольца в более жестких условиях (90°C, 20 атм). Результаты важны для разработки отечественных технологий
переработки растительной биомассы в компоненты моторных топлив.
чл.-корр. РАН В.А. Лихолобов, к.х.н. А.В. Лавренов
Институт проблем переработки углеводородов СО РАН, г. Омск
Влияние природы соединения Мо на физико-химические свойства и активность Mo-содержащего цеолита в процессе неокислительной конверсии метана
Методом твердофазного синтеза с использованием высококремнеземного цеолита с силикатным модулем 40 и различных соединений Мо получены
каталитические системы, исследованы их физико-химические и каталитические свойства в процессе неокислительной конверсии метана в ароматические углеводороды. Для приготовления катализаторов
использовались нанопорошок (НП) Мо (К-1), оксид (К-2) и карбид (К-3) молибдена, молибденовый ангидрид (К-4). Содержание Мо в катализаторах составляло 4,0 % мас.
Введение Мо в цеолит приводит к уменьшению его удельной поверхности и объема пор независимо от соединения Мо. Наиболее заметное снижение удельной
поверхности наблюдается при использовании молибденового ангидрида, что связано с блокировкой каналов цеолита частицами более крупного
размера. Характер изменения кислотности цеолита зависит от природы используемого источника Мо, при этом во всех случаях наблюдается заметное снижение концентрации бренстедовских кислотных центров.
Катализатор с добавкой Мо2С проявил наименьшую активность в процессе конверсии метана, а наиболее высокую активность показал катализатор, содержащий НП Мо. Распределение Мо на поверхности и в каналах цеолита
в этом катализаторе отличается от систем, полученных с использованием других соединений Мо. При прокаливании Мо-содержащего цеолита происходит распределение Мо на его внешней поверхности, при этом
часть Мо мигрирует в каналы. В случае использования НП Мо вероятность проникновения Мо в каналы цеолита в процессе прокаливания выше, чем
при использовании МоО3 и Мо2С. Частицы оксида Мо, расположенные на поверхности цеолита, легко восстанавливаются метаном в ходе реакции до Мо2С, а соединения Мо в каналах
цеолита восстанавливаются медленнее, и могут оставаться частично окисленными, что благоприятно сказывается на активности катализатора
в процессе ароматизации метана.
д.х.н. А.В. Восмериков
Институт химии нефти СО РАН, г. Томск
Исследование влияния дефектов структуры перовскита и феррошпинели сложного состава на их каталитические свойства в процессе окислительной конденсации метана
Эффективность гетерогенно-гомогенного процесса окислительной конденсации метана (ОКМ) определяется структурными дефектами оксидных систем, способными
инициировать образование СН3-радикалов при окислении метана. Изучено влияние дефектов структуры перовскита и феррошпинели сложного состава на их каталитические свойства в процессе ОКМ.
Показано, что каталитическое действие системы SrxGd1-xCoO3-δ (0.5 < x < 0.9) в реакциях ОКМ иглубокого окисления метана существенно зависит от характера
расположения катионов Sr2+/Gd3+ по А-позициям структуры перовскита. Системы с беспорядочным расположением катионов Sr2+/Gd3+
характеризуются случайным распределением кислородных вакансий и увеличенной подвижностью кислорода, что благоприятно для реакции глубокого окисления метана до
СО2. Впервые установлено, что упорядочение катионов Sr2+/Gd3+ приводит к локализации кислородных вакансий, снижению общей активности и количества
слабосвязанного кислорода, что обеспечивает более высокую селективность по С2-продуктам в реакции ОКМ (Chemical Communication. 2014, V. 50, N. 46, p. 6112-6115).
При сопоставлении каталитических свойств, фазового состава и катионного распределения железа по кристаллографическим позициям железосодержащих фаз в стационарном состоянии ферросфер с содержанием
Fe2O3 76-97 мас. % установлено, что при содержании Fe2O3 89 мас. % наблюдается изменение маршрута окисления метана. Для ферросфер с
содержанием Fe2O3 < 88.8 мас. % основным маршрутом превращения метана является глубокое окисление. При содержании Fe2O3 ≥ 89 мас. % резко
увеличивается выход C2-углеводородов на фоне низкого вклада глубокого окисления. Впервые показано, что выход C2-углеводородов коррелирует с количеством дефектов в структуре феррошпинели, которые
представляют собой ионы железа с тетраэдрическим катионом Ca2+ и октаэдрической катионной вакансией среди ближайших соседей (Кинетика и катализ, в печати).
д.х.н., проф. А.Г. Аншиц
Институт химии и химической технологии СО РАН, г. Красноярск
Супероснования как катализаторы контролируемой самоорганизации ацетиленов и кетонов
Под действием супероснований типа гидроксид (алкоксид) щелочного металла/диметилсульфоксид две молекулы ацетилена и две молекулы кетона диастереоселективно самоорганизуются в
7-метилен-6,8-диоксабицикло[3.2.1]октаны и 3-ацил-2-циклопентен-1-олы.
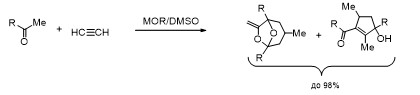
M = Li, Na, K, Cs; R = H, Alk
Соотношение структурных изомеров контролируется концентрацией катализатора и структурой субстрата. Эти каскадные сборки двух перспективных
синтетических строительных блоков и прекурсоров лекарств являются уникальными реакциями образования одновременно трех C-C- и двух C-O- или четырех C-C-связей
из простых реагентов в одном каталитическом процессе.
академик Б.А. Трофимов
Иркутский институт химии СО РАН, г. Иркутск
Разработка кинетических методов установления механизмов сложных каталитических процессов
Предложено семейство кинетических методов установления механизмов сложных каталитических процессов, характеризующихся существенной
нестационарностью активного катализатора, базирующихся не на традиционных измерениях каталитической активности, а на измерениях дифференциальной селективности реакций с искусственно создаваемой
многомаршрутностью. Преимущества предложенных методов обусловлены независимостью дифференциальной селективности от количества активного катализатора при условии, что продукты реакции образуются на одном и
том же катализаторе, что делает интерпретацию экспериментальных данных существенно проще. Условие образования продуктов на одном и том же катализаторе гарантированно выполняется при создании
искусственной многомаршрутности (метод конкурирующих реакций), возникающей при введении в реакцию дополнительных субстратов. Предложен метод исследования дифференциальной селективности по
изотопомерам одного субстрата, не требующий никакой модификации условий проведения реакции. Показано, что визуализация кинетических данных путем построения фазовых траекторий образования продуктов
реакции позволяет оценить изменения дифференциальной селективности катализатора без использования математических описаний, базирующихся на конкретных гипотезах механизма. Полученные результаты
демонстрируют, что измерения дифференциальной селективности позволяют получать информацию о природе активного катализатора, устанавливать скоростьопределяющие стадии каталитического цикла, а также оценивать
степень обратимости его стадий. Кроме того, уравнения дифференциальной селективности имеют более простую форму в сравнении с уравнениями скорости, что является еще одним преимуществом
исследований дифференциальной селективности. Еще одним преимуществом исследований дифференциальной селективности является возможность изучения реакций, проводимых в условиях высоких соотношений
субстрат/катализатор, то есть в реальных каталитических условиях без использования модельных стехиометрических реакций, которые могут приводить к неправильным выводам.
д.х.н. А.Ф. Шмидт
Иркутский государственный университет, г. Иркутск
Установление механизма формирования и природы каталитически активных частиц в димеризации алкенов в присутствии фосфиновых комплексов никеля
Установлен механизм формирования и природа каталитически активных частиц в димеризации алкенов на основе фосфиновых комплексов никеля в
различных степенях окисления в сочетании с эфиратом трифторида бора. Определены каталитические характеристики индивидуальных комплексов Ni(PPh3)2(C2H4) и
Ni(PPh3)nCl (n = 2,3) и систем на их основе в сочетании с кислотами Бренстеда и Льюиса в процессах олигомеризации
этилена и пропилена. Обнаружена связь между временем хранения раствора BF3OEt2 и каталитическими свойствами никелевых систем в превращении низших алкенов. Показано, что
наблюдаемое возрастание частоты и числа оборотов обусловлено увеличением концентрации кислот Бренстеда за счет необратимых превращений BF3OEt2 в результате взаимодействия
с примесями воды в растворителе. Обнаружено резкое повышение длительности функционирования каталитических систем Ni(PPh3)2(C2H4)-BF3OEt2
при формировании их в присутствии субстратов. Комплексом физических методов изучены взаимодействия никелевых прекурсоров с эфиратом трифторида бора и установлены основные реакции формирования
каталитически активных частиц. Показано, что в результате взаимодействия Ni(PPh3)2(C2H4) с эфиратом трифторида бора образуются комплексы Ni(II) с активной
связью Ni-C, которые инициируют процесс ди- и олигомеризации по гидридному механизму. При использовании в качестве прекурсоров фосфиновых комплексов никеля(I) образование в присутствии субстрата и
сокатализатора комплексов Ni(II) с активной связью Ni-C является результатом связывания части фосфиновых лигандов трифторидом бора в аддукты и диспропорционирования комплексов Ni(I) до комплексов Ni(0)
и Ni(II). Сложность исследования процессов формирования и функционирования данных металлокомплексных катализаторов обусловлена тем, что при рассмотрении свойств систем, состоящих из комплексов
никеля в различных степенях окисления (Ni(0), Ni(I), Ni(II)) и алюминий- и борсодержащих компонентов, следует учитывать как кислотно-основные, так и окислительно-восстановительные свойства
последних. Алюминийалкилгалогениды и галогениды бора способны окислять фосфиновые комплексы Ni(0). Кроме того, Al- и B-содержащие сокатализаторы необходимо рассматривать не только как кислоты Льюиса,
но и как исходные вещества при формировании кислот Бренстеда, которые могут образоваться из-за наличия примесей воды в условиях реального каталитического процесса. Решение данных проблем представляет не
только теоретический, но и практический интерес для создания рациональной стратегии поиска новых высокоэффективных катализаторов и оптимизации существующих каталитических систем.
д.х.н. Ф.К. Шмидт, д.х.н. А.Ф. Шмидт
Иркутский государственный университет, г. Иркутск
Разработка способа получения N,N-диэтилокта-2,7-диен-1-амина
Разработан способ получения N,N-диэтилокта-2,7-диен-1-амина, используемого в качестве полупродукта для синтеза ПАВ, антиоксидантов резины,
антикоррозионных добавок. Способ осуществляют путем каталитической теломеризации бутадиена (БД) с диэтиламином в присутствии катализатора на основе катионных комплексов палладия (II),
характеризующимся тем, что в качестве катионных комплексов палладия используют комплексы общей формулы [(acac)Pd(L)2]BF4 (где acac – ацетилацетонатный лиганд, L = PPh3,
PiPr3, PnBu3, P(p-Tol)3 или (L)2 = дифосфиновые лиганды бис(дифенилфосфино)метан, dppm, бис(дифенилфосфино)пропан, dppp, бис(дифенилфосфино)бутан, dppb,
бис(дифенилфосфино) ферроцен, dppf. Предложенный способ позволяет с высокой эффективностью получать N,N-диэтилокта-2,7-диен-1-амин с селективностью 99.9% от общей смеси
продуктов реакции и выходом целевого продукта до TOFav = 1940 ч–1 и TON = 17,480 мольБД/мольPd.
д.х.н. Ф.К. Шмидт, д.х.н. А.Ф. Шмидт
Иркутский государственный университет, г. Иркутск
Палладиевые катализаторы на основе катионных ацетилацетонатных комплексов
Впервые проведен синтез новой серии каталитических прекурсоров — катионных ацетилацетонатных комплексов палладия состава [(acac)Pd(N^N)]BF4 , где N^N – α-дииминовые лиганды. В
качестве дииминовых лигандов были использованы бис[N-(фенил)имино]аценафтен, бис[N-(2,6-диметилфенил)имино]аценафтен, бис[N-(2,6-диизопропилфенил)имино]аценафтен,
N,N-(этандиилиден)бис(2,6-диметиланилин), N,N-(этандиилиден)бис(2,6-диизопропиланилин), N,N-(2,3-бутандиилиден)бис(анилин), N,N-(1,2-дифенилэтан-1,2-диилиден)бис(2,6-диметиланилин),
N,N-(1,2-дифенилэтан-1,2-диилиден)бис(2,6-диизопропиланилин). Каталитические системы на основе комплексов палладия и никеля с α-дииминовыми лигандами, так называемые катализаторы
Брукхарта, хорошо известны в практике современного лабораторного и промышленного гомогенного катализа олиго- и полимеризации низших олефинов. Преимуществом данных каталитических систем является их
относительная устойчивость по отношению к полярным функциональным группам в мономере, а также возможность получения «суперразветвленных» полиолефинов. Основные преимущества
нового класса разработанных палладиевых катализаторов на основе катионных комплексов палладия, обуславливающие их конкурентноспособность: высокая технологичность и относительно низкая
себестоимость синтеза прекурсоров, низкая чувствительность к присутствию в реакционной среде следовых количеств H2O и O2.
д.х.н. Ф.К. Шмидт, д.х.н. А.Ф. Шмидт
Иркутский государственный университет, г. Иркутск
Повышение чувствительности метода магнитно-резонансной томографии газов на основе каталитического гидрирования непредельных соединений параводородом
Актуальность исследований обусловлена важностью развития методов МРТ и их приложений как в фундаментальных физико-химических исследованиях (включая химическую технологию и материаловедение), так и в
современных биомедиицнских исследованиях и практической медицине. Одним из главных недостатков ЯМР и МРТ является относительно невысокая чувствительность. Поэтому для осуществления качественного
прорыва в таких приложениях крайне актуальным и востребованным является развитие методов усиления сигнала с использованием принципов гиперполяризации ядерных спинов. Особо остро проблема
чувствительности стоит для газов из-за существенно более низкой концентрации молекул по сравнению с жидкостями.
Осуществлено гетерогенное каталитическое гидрирование пропилена параводородом на катализаторе Rh/TiO2, что позволило получить поляризацию ядерных спинов пропана на уровне
1,3%, соответствующую усилению сигнала ЯМР 1Н около 810 раз. На основе этого в работе впервые получены трехмерные МРТ изображения протон-содержащих газов с высоким пространственным разрешением
(625×625×625 мкм3) и коротким временем регистрации изображения (< 20 c). При этом за счет усиления сигнала на основе использования новейших методов
гиперполяризации получаемое изображение по качеству не уступает МРТ изображению, полученному по сигналу воды, несмотря на значительно меньшую концентрацию протонов в газе по сравнению с жидкостью.
Использование процесса гетерогенного каталитического гидрирования пропилена параводородом позволяет реализовать простой и дешевый способ получения гиперполяризованных газообразных контрастных агентов
для МРТ.
Все основные результаты получены впервые. Они открывают новые возможности как в материаловедении и химической технологии для визуализации газов в пустотах и каналах (например, в пористых материалах и в реакторах),
так и в биомедицинских приложениях при использовании гиперполяризованных протон-содержащих газов в качестве контрастных агентов для функциональной визуализации легких как альтернатива более
сложным и дорогостоящим исследованиям на основе использования благородных газов (129Хе, 3Не).
Исследования выполнены сотрудниками МТЦ СО РАН, ИК им. Г.К. Борескова СО РАН и НГУ совместно с партнерами из Института томографических исследований
Университета Вандербильта, США (Vanderbilt University Institute of Imaging Science, Nashville, TN, USA).
Результаты работы опубликованы в следующей статье: K.V. Kovtunov, D.A. Barskiy, A.M. Coffey, M.L. Truong, O.G. Salnikov, A.K. Khudorozhkov, E.A. Inozemtseva, I.P. Prosvirin, V.I. Bukhtiyarov,
K.W. Waddell, E.Y. Chekmenev, I.V. Koptyug. High-resolution 3D proton MRI of hyperpolarized gas enabled by parahydrogen and Rh/TiO2 heterogeneous catalyst, Chem. Eur. J., 20, 11636-11639 (2014).
д.х.н. И.В. Коптюг, к.х.н. К.В. Ковтунов, Д.А. Барский, А.В. Сальников, к.х.н. И.П.Просвирин, чл.-корр. РАН В.И. Бухтияров
Международный томографический центр СО РАН,
Институт катализа им. Г.К. Борескова СО РАН,
Новосибирский государственный университет, г. Новосибирск
Разработка новых методов усиления сигнала ЯМР за счет использования параводорода
Целью выполненных исследований является разработка новых методов усиления сигнала ЯМР за счет использования параводорода для создания нового высокочувствительного инструментария ЯМР и МРТ и его применения для
исследования механизмов каталитических реакций и для изучения процессов в функционирующих реакторах, а также для биомедицинских приложений.
Актуальность исследований обусловлена важностью процессов гидрирования непредельных соединений в современной химической промышленности, которые преимущественно
используют катализаторы на основе переходных металлов. Стремление снизить стоимость катализаторов и их воздействие на окружающую среду определяет интерес к созданию новых каталитических систем для
процессов гидрирования на основе более распространенных и безвредных элементов. В частности, среди систем на основе элементов главных групп значительный интерес представляют фрустрированные Льюисовские
пары, для которых в последнее время продемонстрирована возможность использования в процессах активации Н2 и гидрирования широкого круга непредельных соединений.
В работе впервые показана возможность наблюдения усиления сигнала ЯМР за счет эффекта индуцированной параводородом поляризации ядер (ИППЯ) при активации параводорода с использованием не содержащей металла
системы, представляющей собой фрустрированную Льюисовскую пару анса-аминоборана. Продемонстрировано, что активация параводорода этим «молекулярным пинцетом» приводит к усилению сигнала ЯМР
1Н не только обменивающихся, но и других атомов водорода в структуре соединения, а также сигналов ЯМР атома 11B. Полученные результаты демонстрируют применимость метода ИППЯ для
изучения механизма активации Н2 системами, не содержащими атома металла.
Все основные результаты получены впервые и являются существенным продвижением вперед в области развития методов значительного усиления сигнала ЯМР и применения этих методов для исследования механизмов
важных каталитических процессов.
Исследования выполнены сотрудниками МТЦ СО РАН совместно с финскими коллегами из университета Оулу и университета Аалто (Department of Physics, University of
Oulu, and Department of Materials Science and Engineering, Aalto University, Finland) и поддержаны грантом РФФИ (12-03-00403-a) и грантом Президента РФ (MK-1329.2014.3).
Основные результаты опубликованы в следующей статье: 1. V.V. Zhivonitko, V.-V. Telkki, K. Chernichenko, T.J. Repo, M. Leskela, V. Sumerin, I.V. Koptyug. Tweezers for parahydrogen: a metal-free
probe of non-equilibrium nuclear spin states of H2 molecules, J. Am. Chem. Soc., 136, 598-601 (2014).
к.х.н. В.В. Живонитко, д.х.н. И.В. Коптюг
Международный томографический центр СО РАН, г. Новосибирск
Гомогенные и нанесенные катализаторы полимеризации этилена на основе бис(имино) пиридильных комплексов железа(II)
Исследована кинетика полимеризации этилена в присутствии гомогенных катализаторов на основе бис-иминопиридильных комплексов Fe(II) при варьировании состава тридентатного лиганда L
в бис-иминопиридильных комплексах LFeCl2, природы и состава сокатализатора (МАО, триалкилы алюминия). Найдены составы каталитических систем, позволяющих получать линейный
полиэтилен с молекулярной массой от 1×104 до 2×106 г/моль и регулируемым молекулярно-массовым распределением. Методами ЯМР 1Н
и 2Н изучены механизм активации и структура интермедиатов, образующихся при взаимодействии LFeCl2 с различными активаторами (МАО, триалкилы алюминия) в процессе формирования
активного компонента этих систем.
С использованием полученных для гомогенных систем данных разработаны нанесенные катализаторы LFeCl2/SiO2, имеющие высокую и стабильную активность при полимеризации этилена с
сокатализатором – триалкилом алюминия при реальных для технологии температурах (80-90°C) и позволяющие получать линейный полиэтилен с различной молекулярной массой. Эти результаты послужили основой для разработки оригинального
нанесенного бикомпонентного катализатора для производства ПЭ с бимодальным молекулярно-массовым распределением газофазным методом. Новый нанесенный катализатор содержит в своем составе комплекс железа
(LFeCl2), на котором образуется низкомолекулярный линейный полиэтилен, и соединение хрома, на котором образуется разветвленный высокомолекулярный полиэтилен. Результаты испытаний этого
катализатора в НТЦ ОАО «Казаньоргсинтез» подтвердили возможность получения на этом катализаторе бимодального полиэтилена типа ПЭ-100, получаемого в настоящее время на ОАО «Казаньоргсинтез»
с использованием катализатора, закупаемого по импорту.
д.х.н. В.А. Захаров, к.х.н. Н.В. Семиколенова, д.х.н. К.П. Брыляков, к.х.н. И.Е. Сошников, д.х.н. Е.П. Талзи, к.х.н. М.А. Мацько
Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск
Фотокаталитическое окисление дибутилсульфида на фуллеренах. Фотокаталитическое окисление фосфорорганических загрязнителей воды
Работа велась по трём основным направлениям: моделирование наночастиц и их взаимодействия с молекулами, фотокаталитическое окисление
дибутилсульфида на фуллеренах с возможными применениями в тонком органическом синтезе, фотокаталитическое окисление фосфорорганических загрязнителей воды.
Разработаны новые кластерные модели наночастиц диоксида титана в кристаллической модификации анатаз с релаксированными атомами. Разработанные модели и
подходы открывают новые перспективы в компьютерном описании процессов адсорбции и катализа с участием твердых наночастиц.
Исследуется кинетика фотокаталитического окисления органического сульфида на фуллеренах под видимым светом. Идентифицированы основные продукты
реакции, выяснено влияние концентрации начального вещества и интенсивности облучения на скорость и селективность реакции. Использованный метод может быть использован для селективного
парциального окисления органических веществ таких как сульфиды и алкены с использованием солнечного или искусственного света.
Обнаружена повышенная эффективность фотокатализаторов, содержащих гетерополикислоту, адсорбированную на поверхности диоксида титана, в реакциях глубокого окисления фосфорорганических загрязнителей воды.
Фотокатализаторы могут использоваться для разработки устройств тонкой очистки воды.
д.х.н. А.В. Воронцов
Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск
Научные основы синтеза оксидных наноматериалов с заданными свойствами и гетерогенных катализаторов на их основе. Катализаторы дегидратации глицерина
В настоящее время одним из важных аспектов современных химических процессов становится преобразование биомассы и сырья, полученного из
биомассы, в ценные химические вещества и топливо; в частности, переэтерификация растительного масла позволяет получать биодизель и глицерин в качестве побочного продукта. Между тем, глицерин,
содержащий три гидроксильные группы, способен претерпевать химические превращения, обеспечивая получение ценных продуктов. Известно, что дегидратация глицерина в присутствии кислых поверхностных центров
приводит к образованию акролеина, а в присутствии основных – молочной кислоты.
На основе предложенного подхода, основанного на модифицировании двойных слоистых ZnAl-гидроксидов с соотношением Zn/Al ≥ 2 как путем замены межслоевого
аниона NO3– на An– {An–
= [H2W12O40]6–,
WO42–,
HPO42},
так и нанесением РW- или SiW-ГПК 12 ряда, получены композиции, представляющие собой слоистые Zn-Al-гидроксиды со структурой, подобной гидроталькиту. Термическая обработка
модифицированных образцов в интервале 450-600°C приводит к формированию твердого раствора на основе ZnO, а для образцов с нанесенными РW(SiW)-ГПК появляется дополнительная фаза ГПК в высокодисперсном состоянии. На
поверхности рассмотренных образцов присутствуют как бренстедовские (БКЦ), так и льюисовские (ЛКЦ) кислотные центры, причем концентрация БКЦ увеличивается при модифицировании ZnAl-гидроксидов
как при замене межслоевого аниона, так и при нанесении ГПК, а ЛКЦ, наоборот, уменьшается. Показано, что основными продуктами газофазной
дегидратации глицерина на указанных образцах являются акролеин и гидроксиацетон (ацетол); доля других продуктов невелика. Активность
катализаторов увеличивается с повышением температуры реакции (Тр) и при Тр = 325°C конверсия глицерина в зависимости от природы катализатора изменяется
в интервале 30 ÷ 99%. Максимальная селективность по ацетолу (~74%) наблюдается на немодифицированном ZnAl-образце, в то время, как по акролеину (~56%) – на модифицированных
SiW/ZnAl-образцах. В свою очередь, выход акролеина коррелирует с концентрацией БКЦ, а выход ацетола – с концентрацией ЛКЦ. Полученные зависимости согласуются с литературными
данными, согласно которым БКЦ являются ответственными за образование акролеина.
д.х.н. А.С. Иванова
Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск
Научные основы синтеза оксидных наноматериалов с заданными свойствами и гетерогенных катализаторов на их основе. Катализаторы дегидрирования изобутана
Традиционным промышленным способом получения изобутилена является дегидрирование изобутана в кипящем слое с использованием циркулирующего
мелкосферического алюмохромового катализатора. Дегидрирование проводят чередующимися циклами дегидрирование – регенерация. Высокие температуры, попеременное пребывание катализатора в
окислительной и восстановительной средах и непрерывная циркуляция определяют повышенные требования к таким эксплуатационным показателям как механическая прочность гранул и невысокая абразивность. По
эксплуатационным показателям в процессе дегидрирования изобутана наиболее соответствуют микросферические алюмохромовые катализаторы (КДМ, КДМ-М), производимые в промышленном масштабе на OOO
«НПК Синтез», недостатком которых является относительно невысокая селективность (93-94%) по изобутилену.
В лаборатории приготовления катализаторов совместно с лабораторией нестационарных каталитических методов очистки газов разработана
технология получения усовершенствованного микросферического алюмохромового катализатора, обеспечивающего более высокую селективность (96-97%) по изобутилену при сохранении требуемых
эксплуатационных характеристик. Усовершенствование микросферического алюмохромового катализатора проведено на основе алюмооксидных носителях, производимых на OOO «НПК Синтез».
д.х.н. А.С. Иванова, д.х.н. Г.А. Зенковец
Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск
Новая технология получения сверхвысокомолекулярного полиэтилена
В Cанкт-Петербургском филиале ИК СО РАН впервые разработана новая технология получения сверхвысокомолекулярного полиэтилена (СВМПЭ) с
использованием особого класса моноцентровых постметаллоценовых каталитических систем, которая обеспечивает улучшенную морфологию СВМПЭ и его способность перерабатываться при температурах ниже
температуры плавления (из твердой фазы) в сверхпрочные и сверхмодульные изделия (волокна, пленки и т.д.).
Перерабатываемость, высокие прочностные и модульные характеристики полученных в СПб филиале ИК СО РАН образцов реакторных порошков (РП) СВМПЭ были
подтверждены в Институте синтетических полимерных материалов им. Н.С. Ениколопова РАН.
Новая технология отличается от применяемой в настоящее время в мировой практике переработки РП СВМПЭ в изделия из раствора методом гель-формования через фильеры высокой производительностью, упрощённым
аппаратурным оформлением и низкими энергетическими затратами.
По разработанной технологии синтеза СВМПЭ получен патент РФ № 2459835С2 с приоритетом от 18.11.2010 г. (опубликован 27.05.2012 г.) и подана заявка №2013124441 от 24.07.2013 г. на патент РФ, которая находится
на рассмотрении в Федеральном институте промышленной собственности.
чл.-корр. РАН С.С Иванчев
Санкт-Петербургский филиал Института катализа им. Г.К. Борескова СО РАН, г. Санкт-Петербург
Эпоксидирование этилена поверхностным анион-радикальным α-кислородом
Впервые исследовано эпоксидирование этилена поверхностным анион-радикальным α-кислородом, образующимся на цеолите FeZSM-5. Реакция С2Н4
+ Оα протекает без отрыва водорода и ведет к селективному присоединению Оα по двойной связи С=С, с образованием этиленоксида в качестве первичного продукта. Модификация
образца натрием приводит к значительному увеличению селективности по этиленоксиду, достигающей 80-84% при температуре реакции –60°C. Способность α-кислорода,
обладающего высокой электрофильностью, вести селективное эпоксидирование может служить дополнительным аргументом в пользу теории о ключевой роли электрофильного кислорода в эпоксидировании
этилена на Ag-содержащих катализаторах.
На поверхности FeZSM-5, модифицированного Na, обнаружена ранее не известная реакция – взаимодействие этиленоксида с этиленом с образованием бутаналя:
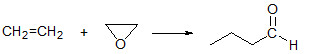
Результаты работы опубликованы в следующей статье: Eugeny V. Starokon, Mikhail V. Parfenov, Larisa V. Pirutko, Igor E. Soshnikov, Gennady I. Panov.
Epoxidation of ethylene by anion radicals of α-oxygen on the surface of FeZSM-5 zeolite. Journal of Catalysis, Vol. 309, January 2014, p. 453-459.
д.х.н. А.С. Харитонов, к.х.н. Е.В. Староконь, М.В. Парфенов,
к.х.н. Л.В. Пирютко, к.х.н. И.Е. Сошников, д.х.н., проф. Г.И. Панов
Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск
Разработка и усовершенствование промышленных катализаторов и технологий
Получение синтетических жидких углеводородов из природного газа
Проведена разработка второй опытной установки полного цикла с реакторами промышленного масштаба по получению синтетических жидких
углеводородов из природного газа в интересах ООО «ИНФРА». Установка построена, испытана и принята в эксплуатацию в ООО «ИНФРА».
Проведены успешные исследования механизма реакций на композиционных цеолитных катализаторах синтеза Фишера-Тропша. Установлено, что карбкатионный
механизм играет решающую роль в составном процессе. По результатам исследования опубликован обзор в журнале «Успехи химии».
Установлено влияние введения различных металлов (алюминий, медь, цинк, железо) в композитные цеолитсодержащие катализаторы синтеза Фишера-Тропша.
Разработаны исходные данные для строительства катализаторной фабрики в интересах ООО «ИНФРА».
д.х.н. В.З. Мордкович
Технологический институт сверхтвердых и новых углеродных материалов, г. Троицк, г. Москва
Катализатор и процесс синтеза дивинила из этанола; катализатор получения пропилена дегидрированием пропана
Продолжены исследования оригинального процесса и катализатора синтеза бутадиена-1,3 из возобновляемых ресурсов – этанола или смеси
этанола и ацетальдегида. По технологии ОАО НИИ «Ярсинтез» выход дивинила на пропущенный этанол приблизительно в два раза выше при селективности по дивинилу на ~ 20% отн. выше, чем в
классическом процессе. Основное внимание уделялось практическим вопросам реализации процесса. В настоящее время заканчивается монтаж опытной установки для подготовки данных на проектирование
промышленного процесса.
Проведены работы по изучению стабильности химического и фазового состава, каталитических и физико-химических характеристик нового
высокоэффективного катализатора в процессе дегидрирования пропана. Установлено, что в течение 1000 ч работы катализатора в реакции дегидрирования не происходит снижения его не происходит снижения его
каталитической активности. Заключено лицензионное соглашение на процесс дегидрирования и производство катализатора.
д.т.н., проф. Г.Р. Котельников
ОАО НИИ «Ярсинтез», г. Ярославль
Исследование физико-химических свойств частично отработанного платинорениевого катализатора риформинга
Каталитический риформинг является одним из базовых процессов переработки нефти. Уровень технологии этого процесса, техническая и экономическая
эффективность в существенной степени зависят от свойств применяемого катализатора. Наибольшей надежностью в эксплуатации отличаются катализаторы с оптимальным содержанием Pt,
Re, и Cl, нанесённые на носитель – оксид алюминия. В настоящее время в России преобладает технология со стационарным слоем катализаторов, в основном импортного производства.
В институте нефти и газа СФУ г. Красноярска изучены физико-химические свойства частично отработанного катализатора Pt-Re-Cl/Al2O3 риформинга нефтяных фракций
(R-98 UOP), ОАО «Ачинский НПЗ» (компания «Роснефть»).
При исследовании физико-химических свойств платинорениевого катализатора риформинга R-98 установлено, что в ходе его эксплуатации происходило необратимое
изменение структурных свойств: укрупнение кристаллитов активной формы носителя η-Al2O3, его частичное фазовое превращение в корунд. Обнаружение корунда
α-Al2O3 в отработанном катализаторе служит указанием на то, что в реакторе имело место значительное повышение температуры при выжигании углеродистых отложений, что
вызвало фазовый переход активной окиси алюминия η-Al2O3 в неактивную высокотемпературную модификацию корунда.
Установлено, что в ходе эксплуатации происходило уменьшение величины удельной поверхности и некоторое увеличение размера пор. При этом показатели механической прочности мало изменялись.
Важная особенность проб отработанного катализатора связана с повышенным содержанием в них примеси оксида железа, который является ядом для металлического компонента катализатора. Накопление оксида железа
может быть связано с коррозией оборудования под действием агрессивной среды, содержащей пары воды и хлористые соединения.
Присутствие углерода в пробах отработанного катализатора указывает на неполное удаление коксовых отложений на стадии окислительной регенерации, что
может быть вызвано недостаточной длительностью процедуры выжига или нарушениями газового потока в реакторе.
Методом ДСК показано существование различных типов углеродистых отложений на поверхности катализатора R-98. Полное удаление остаточных отложений кокса происходит при температуре
500-550°C.
На основании проведённых исследований разработаны рекомендации по увеличению эффективности и продолжительности срока службы используемого катализатора.
Подробные результаты работы опубликованы в Журнале Сибирского федерального университета. Техника и технологии, 2014, том 7, № 8, с. 919-928.
д.т.н. Н.Н. Довженко, д.х.н. В.П. Твердохлебов, П.Н. Кузнецов, А.В. Казбанова, Л.И. Кузнецова, Л.С. Тарасова
Сибирский Федеральный университет, Институт нефти и газа, г. Красноярск
Разработка катализаторов для гидроочистки пропана
Для промышленной очистки пропана от метанола, диметилового эфира в среде циркулирующего водородсодержащего газа используется дорогостоящий
алюмоплатиновый катализатор. Процесс осуществляется при температурах до 370°С, которые позволяют очистить пропан не только от метанола, диметилового эфира, непредельных углеводородов, но и от
органических соединений серы.
В «НИАП-КАТАЛИЗАТОР» (г. Новомосковск) на основе ряда промышленных катализаторов разработаны модифицированные образцы, которые прошли длительные испытания в процессе очистки пропана в
заводской лаборатории гидрирования углеводородов. Предлагаемые катализаторы значительно дешевле используемого драгметального контакта.
Результаты исследования эксплуатационных качеств разработанных никелевых, никельмедных, никельмедьмарганцевых алюмокальциевых модифицированных катализаторов сравнивались с результатами испытаний алюмоплатинового,
никелькизельгурового, никельхромового и других катализаторов. Испытания активности проводили при объемной скорости 200 – 500 ч–1, давлении 8 – 20 кгс/см2,
температурах 180 – 380°С. Перед испытаниями проводилась активация катализаторов в токе водорода при различных температурах.
Проведенные исследования одного из разработанных катализаторов показали ряд существенных достоинств применительно к процессу гидроочистки пропана. Катализатор стабильно работает, не теряя каталитической
активности. На этом этапе время испытаний составляло более 2 суток. На данном катализаторе происходит и гидрирование диоксида углерода. В отличие от других контактов, где содержание СО и СО2
увеличивалось, концентрация СО2 уменьшалась, а образование СО не отмечалось, что является положительным фактором для данного процесса. На синтезированных катализаторах не отмечено значительное
образование тяжелых углеводородов С5 и выше. На данном катализаторе удалось значительно снизить температуру процесса.
Марка
катализатора |
Никель
кизельгуровый |
Алюмо-
платиновый |
Никель-
хромовый |
новый контакт
"НИАП-КАТАЛИЗАТОР" |
|
Температура процесса |
320 – 380 °C
|
260 – 280 °C |
200 – 240 °C |
150 – 190 °C |
К настоящему времени из серии разработанных контактов пока прошли испытания два катализатора. Начата отработка режима снижения температуры процесса активации, приемлемого для промышленной эксплуатации катализаторов.
д.х.н., проф. Е.З. Голосман
«НИАП-КАТАЛИЗАТОР», г. Новомосковск
Синтез метилхлорида окислительным хлорированием метана при повышенном давлении
Для процесса получения легких олефинов из природного газа через промежуточный синтез и пиролиз хлористого метила проводились
исследования по синтезу хлористого метила методом каталитического окислительного хлорирования метана. Для эффективного съема тепла
реакции и увеличения селективности по хлористому метилу в сумме хлорметанов не менее 90-92% процесс оксихлорирования метана проводится с ~
пятикратным по отношению к хлору избытком метана.
В целях увеличения единичной производительности реакторов окислительного хлорирования метана проведены исследования процесса
при давлении до 10 ата.
Экспериментально показана возможность увеличения производительности реактора оксихлорирования метана за счет повышения давления с коэффициентом
пропорциональности ~ Р0.85. Селективность образования метилхлорида при этом не изменяется.
Полученные результаты могут быть использованы при разработке технологии промышленного процесса получения низших олефинов из природного газа
через промежуточный синтез метилхлорида.
д.х.н., проф. Ю.А. Трегер, к.х.н. В.Н. Розанов
Научно-исследовательский инженерный центр «Синтез», г. Москва
Окислительное хлорирование этилена на высокотемпературных катализаторах
Использование высокотемпературных (240-260°C) катализаторов в процессе окислительного хлорирования этилена в псевдоожиженном слое позволяет в 1,5-2 раза повысить
производительность действующих промышленных реакторов, что является составной частью увеличения мощности производств винилхлорида по сбалансированной схеме. Увеличение производительности реакторов
достигается за счет увеличения Δt между слоем катализатора и системой теплосъема.
По сравнению с низкотемпературными (210-225°C) высокотемпературные катализаторы в этих условиях обладают пониженной активностью (константа скорости при 220°C
ниже в 4-5 раз). Компенсация достигается путем повышения температуры процесса.
Снижение активности катализатора с одновременным повышением селективности образования целевого 1,2-дихлорэтана достигается путем ввода в катализатор наряду с хлоридом меди щелочных/щелочноземельных
металлов.
В результате проведенных исследований показано, что наличие в системе MgCl2 способствует стабилизации активных комплексов на поверхности зерна и ведет к повышению
эффективности использования этилена в «воздушном» процессе до 95-96%; выход продуктов глубокого окисления при 240оС не превышает 1,5-1,8%.
Использование в качестве стабилизирующей добавки хлорида лития позволяет получить схожие результаты, хотя показатели процесса при 240-250°C
несколько уступают системам, содержащим хлорид магния.
Показано, что смесь катализаторов дает результаты, близкие к системам, содержащим CuCl2-MgCl2/Al2O3.
д.т.н. М.Р. Флид
Научно-исследовательский инженерный центр «Синтез», г. Москва
Получение низших олефинов пиролизом метилхлорида
Для процесса получения низших олефинов из природного газа через промежуточный синтез и пиролиз хлористого метила проведены широкие
поисковые исследования по подбору аналога силикоалюмофосфатного катализатора SAPO-34. Были использованы различные исходные вещества и условия синтеза, различные связующие.
Определены оптимальные физико-химические характеристики катализатора, позволяющие достичь степени превращения метилхлорида более 70% за
проход при суммарной селективности образования этилена и пропилена более 80%.
Обнаружено, что селективность процесса пиролиза хлористого метила до низших олефинов значительно возрастает и достигает максимума при
определенной степени зауглероживания катализатора, в то время как активность катализатора монотонно падает по мере зауглероживания.
Показано, что после регенерации воздухом активность катализатора полностью восстанавливается.
д.х.н., проф. Ю.А. Трегер, к.х.н. В.Н. Розанов
Научно-исследовательский инженерный центр «Синтез», г. Москва
Разработка технологии и производство партии кобальт-фталоцианинового катализатора ИВКАЗ
Разработана технология производства кобальт-фталоцианинового катализатора ИВКАЗ в жидкой форме (20%-ый водно-щелочной раствор) для процессов очистки
нефтей (ДМС) и нефтепродуктов (ДМД). Произведено и поставлено российским и зарубежным заказчикам 2500 кг катализатора ИВКАЗ.
академик АН РТ, Генеральный директор, д.т.н. А.М. Мазгаров
ОАО Волжский научно-исследовательский институт углеводородного сырья, г. Казань
Разработка катализаторов изомеризации и усовершенствование технологии производства
В процессе разработки катализатора изомеризации С7-фракции для снижения ароматических углеводородов в товарных автобензинах
проведены пилотные испытания опытно-промышленной партии катализатора СИ-4. Подтверждены показатели процесса «Изомалк-4» для промышленного внедрения.
При разработке катализатора изомеризации н-бутана проведены пилотные испытания катализатора СИ-4. По результатам пилотных испытаний разработан базовый проект установки изомеризации н-С4
в компании Shandong Sincier Petrochemical Co., Ltd. (Китай).
Усовершенствована технология промышленного производства катализатора изомеризации пентан-гексановых фракций. Выпущены две промышленные партии катализатора СИ-2 для ЗАО «Роснефть-РНПК» и ОАО
«Орскнефтеоргсинтез».
Генеральный директор, к.х.н. А.Н. Шакун
ОАО НПП «Нефтехим», г. Краснодар
Синтез сорбентов и катализаторов для очистки атмосферы
Проводятся работы по синтезу гидрофобных цеолитов применительно к очистке атмосферы от вредных компонентов. Наиболее актуальной проблемой
является, в частности, каталитическое разложение токсичных компонентов до безвредных веществ в процессе адсорбции. В этой связи
в Корпорации ведутся работы по поиску новых фотокатализаторов на основе титано-силикатных цеолитов. В настоящее время осуществляются работы по синтезу нового класса сорбентов и катализаторов на основе
металл-органических структур для очистки обитаемых помещений от токсичных компонентов.
д.т.н. С.Б. Путин
ОАО «Корпорация «Росхимзащита», г. Тамбов

 Iidazolium-based N-heterocyclic carbenes (NHCs) are a versatile set of catalyst molecules. Chemists use them as ligands in transition-metal
catalysts, and they use them alone as metal-free organocatalysts. They would also like enantioselective versions of these compounds to
prepare chiral molecules, but so far only a few successful versions have been reported.
Iidazolium-based N-heterocyclic carbenes (NHCs) are a versatile set of catalyst molecules. Chemists use them as ligands in transition-metal
catalysts, and they use them alone as metal-free organocatalysts. They would also like enantioselective versions of these compounds to
prepare chiral molecules, but so far only a few successful versions have been reported. By taking cues from two biological processes, researchers have made a catalytic material that converts nitrogen to ammonia when irradiated by white light (J. Am. Chem. Soc. 2015, DOI: 10.1021/ja512491v).
The new strategy may one day help scientists achieve energy savings in various catalytic processes by capitalizing on abundant sunlight to produce valuable chemicals.
By taking cues from two biological processes, researchers have made a catalytic material that converts nitrogen to ammonia when irradiated by white light (J. Am. Chem. Soc. 2015, DOI: 10.1021/ja512491v).
The new strategy may one day help scientists achieve energy savings in various catalytic processes by capitalizing on abundant sunlight to produce valuable chemicals.

 PRESSURE PROCESS
PRESSURE PROCESS

 31 января 2015 года исполнилось 75 лет действительному члену Российской академии наук, декану Химического факультета Московского государственного университета имени М.В. Ломоносова, профессору Валерию Васильевичу Лунину.
31 января 2015 года исполнилось 75 лет действительному члену Российской академии наук, декану Химического факультета Московского государственного университета имени М.В. Ломоносова, профессору Валерию Васильевичу Лунину.