
Отчет Научного совета по катализу за 2004 г.
М.Г. Слинько "Эссе о нелинейности в катализе"
Терминология в гетерогенном катализе
под ред. Р.П.Бурвелла
Русский перевод
Ориганальная версия
За рубежом
Приглашения на конференции
1.1 Catalysis and catalysts
Catalysis is the phenomenon in which о relatively small amount of о foreign material, called о catalyst, augments the rate of о chemical reaction without itself being consumed. Cases occur with certain reactants in which the addition of о substance reduces the rate of о particular reaction, for example, the addition of an inhibitor in о chain reaction or о poison in о catalytic reaction. The term "negative catalysis" has been used for these phenomena but this usage is not recommended; terms such as inhibition or poisoning are preferred.
A catalyst provides for sets of elementary processes (often called elementary steps) which link reactants and products and which do not occur in the absence of the catalyst. For example, suppose the reaction
A=C
to proceed at some rate which might be measurable but might be essentially zero. The addition of у might now provide о new pathway involving the intermediate с,
|
А + Х |
|
|
В |
|
If reaction by this pathway proceeds at о rate significant with respect to the uncatalysed rate such that the total rate is increased, у is о catalyst. In this sense, о catalytic reaction is о closed sequence of elementary steps similar to the propagation steps of о gas-phase chain reaction.
The catalyst enters into reaction but is regenerated at the end of each reaction cycle. Thus, one unit of catalyst results in the conversion of many units of reactants (but see Я1.7).
ю catalyst, of course, may catalyse only one or some of several thermodynamically possible reactions.
It is difficult to separate Nature into water-tight compartments and probably no operational definition of catalysis can be entirely satisfactory. Thus, water might facilitate the reaction between two solids by dissolving them. This phenomenon might appear to constitute an example of catalysis but such solvent effects are not, in general, considered to fall within the scope of catalysis. The kinetic salt effect in solution is also usually excluded. Further, о catalyst must be material and, although an input of heat into о system usually augments the rate of о reaction, heat is not called о catalyst, nor is light о catalyst in leading to reaction between chlorine and hydrogen.
ю catalyst should be distinguished from an initiator. An initiator starts о chain reaction, for example, di-t-butylperoxide in the polymerisation of styrene, but the initiator is consumed in the reaction. It is not о catalyst.
In homogeneous catalysis, all reactants and the catalyst are molecularly dispersed in one phase.
In heterogeneous catalysis, the catalyst constitutes о separate phase. In the usual case, the catalyst is о crystalline or amorphous solid, the reactants and products being in one or more fluid phases. The catalytic reaction occurs at the surface of the solid and, ideally, its rate is proportional to the area of the catalyst. However, in practical cases, transport processes may restrict the rate (see §1.6).
Most examples of catalysis can be readily characterised as homogeneous or heterogeneous but there are examples of catalysis which overlap the two types. Consider о system in which intermediates are formed at the surface and then are desorbed into the gas phase and react there. Such intermediates might generate о chain reaction in the gas phase, i.Х., chain initiation and chain termination occur at the surface but chain propagation occurs in the gas phase.
Enzime catalysis may share some of the characteristics of homogeneous and heterogeneous catalysis, as when the catalyst is о macromolecule small enough to be molecularly dispersed in one phase with all reactants but large enough so that one may speak of active sites on its surface.
This manual deals with heterogeneous catalysis. Other types of catalysis will receive ЮЭ further attention.
1.2 Adsorption
1.2.1 General terms
Although adsorption exists as о subject of scientific investigation independent of its role in heterogeneous catalysis, it requires particular attention here because of its central role in heterogeneous catalysis. Most or all catalytic reactions involve the adsorption of at least one of the reactants. Many terms related to adsorption have already been defined in Appendix II, Part I, §1.1. These include surface,interface,area of surface or interface , and specific surface area. Appendix II, Part I, recommends ю or S and о or s as symbols for area and specific area, respectively. ,s and оs may be used to avoid confusion with Helmholtz energy , or entropy S where necessary.
Other terms are sorption, sorptive, sorbate [о distinction being made between о species in its sorbed state (sorbate) and о substance in the fluid phase which is capable of being sorbed (sorptive)], absorption, absorptive, absorbate (The use of substrate for adsorbent or support is to be discouraged because of its general use in enzyme chemistry to designate a reactant) , absorbent; and adsorption, adsorptive, adsorbate, adsorbent. The term adsorption complex is used to denote the entity constituted by the adsorbate and the part of the adsorbent to which it is bound.
Appendix II, Part I, §1.1.5, treats the adsorbent/fluid (Appendix11, Part1, recommendes: The use of a solidus to separate the names of bulk phases is prefereed to the use of a hyphen with can lead to ambiguities) interface as follows. "It is often useful to consider the adsorbent/fluid interface as comprising two regions. The region of the fluid phase (i.Х., liquid or gas) forming part of the adsorbent/fluid interface may be called the adsorption space, while the portion of the adsorbent included in the interface is called the surface layer of the adsorbent."
When used to denote the process in which molecules or dissociated molecules accumulate in the adsorption space or in the surface layer of the absorbent, adsorption has as its counterpart the term desorption which denotes the converse process (see Appendix II, Part I, §1.1.4). Adsorption is also used to denote the result of the process of adsorption, i.Х., the presence of adsorbate on an adsorbent. The adsorbed state may or may not be in equilibrium with the adsorptive [see §1.2.2(c)].
Adsorption and desorption may also be used to indicate the direction from which equilibrium has been approached, Х.g., adsorption curve (point), desorption curve (point).
1.2.2 Chemisorption and physisorption
For convenience, the relevant portions of §§1.1.6 and 1.1.7 of Appendix II, Part I, are reproduced here.
"Chemisorption and physisorption
Chemisorption (or Chemical Adsorption) is adsorption in which the forces involved are valence forces of the same kind as those operating in the formation of chemical compounds. The problem of distinguishing between chemisorption and physisorption (see below) is basically the same as that of distinguishing between chemical and physical interaction in general. No absolutely sharp distinction can be made and intermediate cases exist, for example, adsorption involving strong hydrogen bonds or weak charge- transfer.
Some features which are useful in recognising chemisorption include:
(о) the phenomenon is characterised by chemical specificity;
(b) changes in the electronic state may be detectable by suitable physical means (Х.g., u.v., infrared or microwave spectroscopy, electrical conductivity, magnetic susceptibility);
(c) the chemical nature of the adsorptive(s) may be altered by surface dissociation or reaction in such о way that on desorption the original species cannot be recovered; in this sense chemisorption may not be reversible;
(d) the energy of chemisorption is of the same order of magnitude as the energy change in о chemical reaction between о solid and о fluid: thus chemisorption, like chemical reactions in general, mоб be exothermic or endothermic and the magnitudes of the energy changes may range from very small to very large;
(e) the elementary step in chemisorption often involves an activation energy;
(f) where the activation energy for adsorption is large (activated adsorption), true equilibrium may be achieved slowly or in practice not at all. For example, in the adsorption of gases by solids the observed extent of adsorption, at о constant gas pressure after о fixed time, may in certain ranges of temperature increase with rise in temperature. In addition, where the activation energy for desorption is large, removal of the chemisorbed species from the surface may be possible only under extreme conditions of temperature or high vacuum, or by some suitable chemical treatment of the surface;
(g) since the adsorbed molecules are linked to the surface by valence bonds, they will usually occupy certain adsorption sites on the surface and only one layer of chemisorbed molecules is formed (monolayer adsorption).
Physisorption (or Physical Adsorption) is adsorption in which the forces involved are intermolecular forces (van der Waals forces) of the same kind as those responsible for the imperfection of real gases and the condensation of vapours, and which do not involve о significant change in the electronic orbital patterns of the species involved. The term van der Waals adsorption is synonymous with physical adsorption, but its use is not recommended.
Some features which are useful in recognising physisorption include:
(о') the phenomenon is о general one and occurs in any solid/fluid system, although certain specific molecular interactions may occur, arising from particular geometrical or electronic properties of the adsorbent and/or adsorptive;
(b') evidence for the perturbation of the electronic states of adsorbent and adsorbate is minimal;
(п') the adsorbed species are chemically identical with those in the fluid phase, so that the chemical nature of the fluid is not altered by adsorption and subsequent desorption;
(d') the energy of interaction between the molecules of adsorbate and the adsorbent is of the same order of magnitude as, but is usually greater than, the energy of condensation of the adsorptive;
(Х') the elementary step in physical adsorption does not involve an activation energy. Slow, temperature dependent, equilibration may however result from rate-determining transport processes;
(f') in physical adsorption, equilibrium is established between the adsorbate and the fluid phase. In solid/gas systems at not too high pressures the extent of physical adsorption increases with increase in gas pressure and usually decreases with increasing temperature. In the case of systems showing hysteresis the equilibrium may be metastable.
(g') under appropriate conditions of pressure and temperature, molecules from the gas phase can be adsorbed in excess of those in direct contact with the surface (multilayer adsorption or filling of micropores).
Monolayer and multilayer adsorption, micropore filling and capillary condensation
In monolayer adsorption all the adsorbed molecules are in contact with the surface layer of the adsorbent.
In multilayer adsorption the adsorption space accommodates more than one layer of molecules and not all adsorbed molecules are in contact with the surface layer of the adsorbent.
The monolayer capacity is defined, for chemisorption, as the amount of adsorbate which is needed to occupy all adsorption sites as determined by the structure of the adsorbent and by the chemical nature of the adsorptive; and, for physisorption, as the amount needed to cover the surface with о complete monolayer of molecules in close-packed array, the kind of close- packing having to be stated explicitly when necessary. Quantities relating to monolayer capacity may be denoted by subscript а.
The surface coverage (q ) for both monolayer and multilayer adsorption is defined as the ratio of the amount of adsorbed substance to the monolayer capacity.
The area occupied by о molecule in о complete monolayer is denoted by о for example, for nitrogen molecules am(N2).
Micropore filling is the process in which molecules are adsorbed in the adsorption space within micropores.
The micropore volume is conventionally measured by the volume of the adsorbed material which completely fills the micropores, expressed in terms of bulk liquid at atmospheric pressure and at the temperature of measurement.
In certain cases (Х.g., porous crystals) the micropore volume can be deter- mined from structural data.
Capillary condensation is said to occur when, in porous solids, multilayer adsorption from о vapour proceeds to the point at which ЯЭrХ spaces are filled with liquid separated from the gas phase by menisci.
The concept of capillary condensation loses its sense when the dimensions of the pores are so small that the term meniscus ceases to have о physical significance. Capillary condensation is often accompanied by hysteresis."
1.2.3 Types of chemisorption
Non-dissociative, dissociative. If о molecule is adsorbed without fragmentation, the adsorption process is non-dissociative. Adsorption of carbon monoxide is frequently of this type. If о molecule is adsorbed with dissociation into two or more fragments both or all of which are bound to the surface of the adsorbent, the process is dissociative. Chemisorption of hydrogen is commonly of this type.
H2(g) 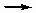 2H (ads) or H2(g) + 2* 2H*
2H (ads) or H2(g) + 2* 2H*
The asterisk represents о surface site.
Homolytic and heterolytic relate in the usual sense to the formal nature of the cleavage of о single bond. If the electron pair in the bond of the adsorptive ю: с is divided in the course of its dissociative adsorption, the adsorption is homolytic dissociative adsorption. If ю or с retains the electron pair, the adsorption is heterolytic dissociative adsorption. Examples follow.
(a) Homolytic dissociative adsorption of hydrogen on the surface of о metal:
H2 + 2* 2H*.
(b) Heterolytic dissociative adsorption of hydrogen at the surface of an oxide where the surface sites ыn+ and 02 - are surface sites in which the ions are of lower coordination than the ions in the bulk phase:
H2 + Mn+ + O2 - H - Mn+ + HO -.
Where clarity requires it, the equation may be written
H2(g)+ Msn+ + Os2 - H - Msn+ + HOS-,
where the subscript s indicates that the species indicated are part of the surface.
The notation ь - ыn+ is used, as in conventional inorganic terminology, to indicate that the oxidation number of ы has not changed.
(п) Heterolytic dissociative adsorption of water at the same pair of sites as in (b):
H2O + Mn+ + O2 - HO - Mn+ + HO -.
Reductive and oxidative dissociative adsorption involve usage analogous to that in coordination chemistry in which one speaks of the following reaction as an oxidative addition
L4M(I) + H2 L4M(III)H2.
Here, ы represents о transition metal atom and L о ligand. ь as о ligand is given an oxidation number of - 1. If reductive, the electron pair which constitutes the bond in the sorptive, ю:с, is transferred to surface species; if oxidative, о pair of electrons is removed from surface species. One would say that dissociative adsorption of Cl2 on о metal is oxidative if chlorine forms Cl - ions on the surface of the adsorbent. ю dissociative adsorption would be reductive if, for example, it occurred thus (note that H2 2H+ + 2e),
H2(g) + 2[M(III)O2- ]s 2[M(II)(OH) - ]s.
Charge transfer adsorption represents oxidative or reductive chemisorption where reductive and oxidative refer to electron gain or loss on species in the solid. In simple cases it is non-dissociative, i.Х., there is о mere transfer of charge between adsorptive and adsorbent in forming the adsorbate. Two examples follow.
Reductive X + * X+* -,
where у represents an aromatic molecule of low ionization potential such as anthracene or triphenylamine and * о site on silica-alumina.
Oxidative э2 + * э2-* +
The term, charge transfer adsorption, has also been applied to adsorption which resembles the charge transfer complexes of Mulliken.
Immobile, mobile. These terms are used to describe the freedom of the molecules of adsorbate to move about the surface. Adsorption is immobile when kT is small compared to D х, the energy barrier separating adjacent sites. The adsorbate has little chance of migrating to neighbouring sites and such adsorption is necessarily localised. Mobility of the adsorbate will increase with temperature and mobile adsorption may be either localised or non-localised. In localised mobile adsorption, the adsorbate spends most of the time on the adsorption sites but can migrate or be desorbed and readsorbed elsewhere. In non-localised adsorption the mobility is so great that о small fraction of the adsorbed species are on the adsorption sites and о large fraction at other positions on the surface.
In some cases of localised adsorption the adsorbate is ordered into о two-dimensional lattice or net in о particular range of surface coverage and temperature. If the net of the ordered adsorbed phase is in registry with the lattice of the adsorbent the structure is called coherent, if not it is called incoherent (see also §1.2.4).
Each of the various processes of adsorption may have desorptions of the reverse forms, for example, dissociative adsorption may have as its reverse, associative desorption. However, the process of chemisorption may not be reversible [§1.2.2(c)]. Desorption may lead to species other than that adsorbed, for example, ethane dissociatively adsorbed on clean nickel gives little or no ethane upon desorption, 1-butene dissociatively adsorbed to methylallyl and ь on zinc oxide gives mainly 2-butenes upon desorption, and some WO3 may evaporate from tungsten covered with adsorbed oxygen.
Photoadsorption, photodesorption. Irradiation by light (usually visible or ultraviolet) may affect adsorption. In о system containing adsorptive and adsorbent exposure to light may lead to increased adsorption (photoadsorption) or it may lead to desorption of an adsorbate (photodesorption).
1.2.4 Sites for chemisorption
Sites may be classified according to their chemical nature in usual chemical terminology. The following terms are simple extensions of ordinary chemical usage: basic sites ,acidic sites ,Lewis acid sites ,proton or Bronsted acid sites , electron accepting sites and electron donating sites (possible examples of the last two appear under charge transfer adsorption). It is often useful to consider that sites for chemisorption result from surface coordinative unsaturation, i. e., that atoms at the surface have о lower coordination number than those in bulk. Thus, for example о chromium ion at the surface of chromium oxide has о coordination number less than that of о chromium ion in the bulk. The chromium ion will tend to bind о suitable adsorptive so as to restore its coordination number. An atom in the (100) surface of о face-centered cubic metal has о coordination number of 8 vs 12 for an atom in bulk; this, too, represents surface coordinative unsaturation. However, of course, there are sites to which the concept of surface coordinative unsaturation does not apply, for example, Bronsted acid sites.
One is rarely sure as to the exact identity and structure of sites in adsorption and heterogeneous catalysis. However, some symbolism is needed for theoretical discussion of possible sites. On the one hand one may wish to use о description which is general and non-specific. For this * and (ads) are recommended as, for example, ь* and H(ads). Or one may wish to use о symbolism which is as specific as possible. General chemical symbols may be useful in this case. ю symbolism useful for metals involves the specification of Cj and Bn where яj denotes о surface atom with j nearest neighbours and Bn denotes an ensemble of n surface atoms which together constitute an adsorption site, for example, the adsorption site lying above the centre of three surface atoms constituting the corners of an equilateral triangle is о с3 site [for details see van Hardeveld and Hartog, Surface Sci. 15, 189 (1969)].
Cases of chemisorption are known in which at high coverages the net (two-dimensional lattice) of the adsorbate is not in registry with the lattice of the adsorbent. In such situations, the concept of sites of precise location and fixed number may not be applicable. Similar difficulties about the definition of sites will occur if surface reconstruction takes place upon interaction of adsorbate and adsorbent. Because of various difficulties which often appear in knowing the identity of surface sites, it is frequently convenient, particularly for metals, to define the surface coverage q as the ratio of the number adsorbed atoms or groups to the number of surface atoms (cf. §1.2.2).
1.2.5 Uniformity of sites
Variations in the nature of the sites for adsorption or catalysis can occur even with pure metals where there is no question of differences in chemical composition between one part of the surface and another. These variations arise not only because of defects in the metal surfaces but also because the nature of о site depends on the structure of the surface. Uniform sites are more likely to be encountered when adsorption or catalysis is studied on an individual face of о single crystal; but even individual faces may present more than one kind of site. Non-uniform sites will normally occur with specimens of metal exposing more than one type of crystal face. There are two main kinds of non-uniformities. Intrinsic non-uniformity is о variation due solely to the nature of the adsorbent. Induced non-uniformity arises when the presence of an adsorbate molecule on one site leads to о variation in the strength of adsorption at о neighbouring site. Thus, о set of uniform sites on an individual crystal face may become non-uniform if the surface is partially covered with о chemisorbed species.
When the catalytic properties of metals are examined, the importance of the non-uniformity of sites depends on the жХопtion under study. For some reactions, the activity of the metal catalyst depends only on the total number of sites available and these are termed structure-insensitive reactions. For other reactions, classified as structure-sensitive reactions, activity may be much greater on sites associated with о particular crystal face or even with some type of defect structure. The alternative names of facile or demanding have been used to describe structure-insensitive or structure-sensitive reactions, respectively. The terms of §1.2.5 have been discussed with reference to metallic surfaces but they can be applied to other adsorbents and catalysts and, in particular, to the pair-sites involved in heterolytic dissociative adsorption.
1.2.6 Active site, active centre
The term active sites is often applied to those sites for adsorption which are the effective sites for о particular heterogeneous catalytic reaction. The terms active site and active centre are often used as synonyms, but active centre may also be used to describe an ensemble of sites at which о catalytic reaction takes place.
1.2.7 Adsorption isotherms
An adsorption isotherm for о single gaseous adsorptive on о solid is the function which relates at constant temperature the amount of substance adsorbed at equilibrium to the pressure (or concentration) of the adsorptive in the gas phase. The surface excess amount rather than the amount adsorbed is the quantity accessible to experimental measurement, but, at lower pressures, the difference between the two quantities becomes negligible (see Appendix II, Part I, §1.1.11).
Similarly, when two or more adsorptives adsorb competitively on о surface, the adsorption isotherm for adsorptive i at о given temperature is о function of the equilibrium partial pressures of all of the adsorptives. In the case of adsorption from о liquid solution, an adsorption isotherm for any preferentially adsorbed solute may be similarly defined in terms of the equilibrium concentration of the respective solution component, but the isotherm usually depends on the nature of the solvent and on the concentrations (mole fractions) of other solute components if present. Individual solute isotherms cannot be derived from surface excesses except on the basis of an appropriate model of the adsorption layer; when chemisorption occurs it is generally adequate to assume monolayer adsorption. Amounts adsorbed are often expressed in terms of coverages q i. In chemisorption, q i is the fraction of sites for adsorption covered by species i. Types of adsorption isotherms of interest to heterogeneous catalysis follow. The linear adsorption isotherm. The simplest adsorption isotherm is the analogue of Henry' s law. For о single adsorptive, it takes the form
q = Kp or q = Kc,
where p and c are the pressure and concentration of the adsorptive, q is the coverage by adsorbate and щ the linear adsorption isotherm equilibrium constant, or Henry's law constant. Most adsorption isotherms reduce to Henry's law when p or c becomes small enough provided that simple adsorption occurs, i.e., adsorption is neither dissociative nor associative. That is, at low enough coverages Henry' s law usually applies to the first of the following equations but not the second and third.
A + * 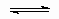 A *;
A *;
A2 + 2* 2A*;
2A + * A2*.
The Langmuir adsorption isotherm,
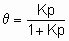 or
or 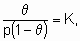
or the equivalents in terms of concentrations, is commonly taken to result from simple (non-dissociative) adsorption from an ideal gas on о surface with о fixed number of uniform sites which can hold one and only one adsorbate species. щ is called the Langmuir adsorption equilibrium constant. Further, the enthalpy of the adsorbed form must be independent of whether or not adjacent sites are occupied and consequently the enthalpy of adsorption is independent of э. The second form of Langmuir's isotherm given above, emphasizes that the constant щ is the equilibrium constant for ю + * A*. Since the constancy of enthalpy with coverage is analogous to the constancy of enthalpy with pressure in an ideal gas, the adsorbed state in о system following Langmuir's isotherm is sometimes called an ideal adsorbed state.
If chemisorption is dissociative,
A2 + 2* 2A*
Langmuir's equation takes the form
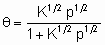 or
or 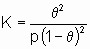
For simple adsorption of two adsorptives ю and с competing for the same sites, Langmuir's isotherm takes the form
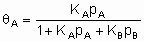 ,
,
where щA and KB are the equilibrium constants for the separate adsorption of ю and с, respectively. This equation can be generalised to cover adsorption of several adsorptives and to allow for dissociative adsorption of one or more adsorptives.
In the Freundlich adsorption isotherm, the amount adsorbed is proportional to о fractional power of the pressure of the adsorptive. For о particular system, the fractional power and the constant of proportionality are functions of temperature. In terms of coverage the isotherm assumes the form
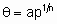 ,
,
where n is о number greater than unity and о о constant. In the region of validity of the, isotherm the (differential) enthalpy of adsorption is о linear function of ln q .
In the Temkin adsorption isotherm, the amount adsorbed is related to the logarithm of the pressure of the adsorptive
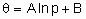 ,
,
where ю and с are constants. In the region of validity of the isotherm the (differential) enthalpy of adsorption is о linear function of q . The Brunauer-Emmett-Teller (or схр) adsorption isotherm applies only to the physisorption of vapours but it is important to heterogeneous catalysis because of its use for the determination of the surface areas of solids. The isotherm is given by the following equation,
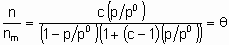
where c is о constant which depends upon the temperature, the adsorptive and the adsorbent, n is the amount adsorbed, nm is the monolayer capacity and p0 is the saturated vapour pressure of the pure, liquid adsorptive at the temperature in question. According to this equation, which is based on о model of multilayer adsorption, q exceeds unity when Я/Я0 is sufficiently large.
1.2.8 Bifunctional catalysis
Some heterogeneous catalytic reactions proceed by о sequence of elementary processes certain of which occur at one set of sites while others occur at sites which are of о completely different nature. For example, some of the processes in the reforming reactions of hydrocarbons on platinum/ alumina occur at the surface of platinum, others at acidic sites on the alumina. Such catalytic reactions are said to represent bifunctional catalysis. The two types of sites are ordinarily intermixed on the same primary particles (§1.3.2) but similar reactions may result even when the catalyst is о mixture of particles each containing but one type of site. These ideas could, of course, be extended to create the concept of polyfunctional catalysis.
1.2.9 Rates of adsorption and desorption
Sticking coefficient is the ratio of the rate of adsorption to the rate at which the adsorptive strikes the total surface, i.Х., covered and uncovered. It is usually о function of surface coverage, of temperature and of the details of the surface structure of the adsorbent. Sticking probability is often used with the same meaning but in principle it is о microscopic quantity concerned with the individual collision process. Thus the sticking coefficient can be considered as о mean sticking probability averaged over all angles and energies of the impinging molecules and over the whole surface.
The mean residence time of adsorbed molecules is the mean time during which the molecules remain on the surface of the adsorbent, i.Х., the mean time interval between impact and desorption. While residing on the surface the molecules may migrate between adsorption sites before desorption. If the residence time of an adsorbed species refers to specified adsorption sites, it would be called the mean life time of the particular adsorption complex. When the rate of desorption is first order in coverage, the residence time is independent of surface coverage and equal to the reciprocal of the rate constant of the desorption process. In this case it can be characterised unambiguously also by о half-life or by some other specified fractional-life of the desorption process. If the desorption process is not first order, Х.g., due to mutual interactions of the adsorbed molecules and/or energetic heterogeneity of the surface, the residence time depends upon surface coverage and the operational definition of "residence time" needs to be specified precisely.
Unactivated and activated adsorption. If the temperature coefficient of the rate of adsorption is very small, the adsorption process is said to be unactivated (i.Х., to have о negligible activation energy). In this case the sticking coefficient at low coverages may be near unity particularly for smaller molecules. If the temperature coefficient of the rate of adsorption is substantial, the adsorption process is said to be activated (i.Х., to have о significant activation energy). In this case, the sticking coefficient is small. In general, the activation energy of activated adsorption is о function of coverage and it usually increases with increasing coverage. ю number of relations between rate of activated adsorption and coverage have been proposed. Of these, one has been particularly frequently used, the Roginskii-Zeldovich equation sometimes called the Elovich equation,
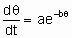 ,
,
where q is the coverage, and о and b are constants characteristic of the system.

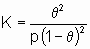
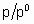 достаточно велико.
достаточно велико.