IV Научная конференция
БОРЕСКОВСКИЕ ЧТЕНИЯ
19-21 апреля 2017 г., Новосибирск, Россия
http://conf.nsc.ru/boreskov-IV/en
 С 19 по 21 апреля в Новосибирском Академгородке прошла IV Конференция БОРЕСКОВСКИЕ ЧТЕНИЯ, посвященная 110-летнему юбилею со дня рождения выдающегося ученого, основателя и первого директора Института катализа, академика Георгия Константиновича Борескова. Для науки и практики катализа основополагающее значение сыграло последовательное развитие Г.К. Боресковым представлений о катализе как о сугубо химическом явлении, в котором решающую роль играют промежуточные химические взаимодействия реактантов и катализаторов.
С 19 по 21 апреля в Новосибирском Академгородке прошла IV Конференция БОРЕСКОВСКИЕ ЧТЕНИЯ, посвященная 110-летнему юбилею со дня рождения выдающегося ученого, основателя и первого директора Института катализа, академика Георгия Константиновича Борескова. Для науки и практики катализа основополагающее значение сыграло последовательное развитие Г.К. Боресковым представлений о катализе как о сугубо химическом явлении, в котором решающую роль играют промежуточные химические взаимодействия реактантов и катализаторов.
IV Конференция проходила в Доме ученых СО РАН и Институте катализа им. Г.К. Борескова СО РАН. Конференция была организована при финансовой поддержке Российского фонда фундаментальных исследований и спонсорской поддержке Партнеров: Фонда инфраструктурных и образовательных программ (РОСНАНО), Honeywell UOP, Института проблем переработки углеводородов СО РАН, АО «СКТБ «Катализатор»» и ООО «Академ-комплект». Организаторами конференции выступили Сибирское отделение РАН, Институт катализа им. Г.К. Борескова СО РАН, Новосибирский государственный университет, Институт органической химии им. Н.Д. Зелинского РАН, Научный совет по катализу ОХНМ РАН, Московский государственный университет имени М.В. Ломоносова, Институт проблем переработки углеводородов СО РАН и Новосибирское отделение Российского химического общества им. Д.И. Менделеева.

Слева направо: научный руководитель ИК СО РАН, академик РАН Валентин Николаевич Пармон,
директор ИК СО РАН, академик РАН Валерий Иванович Бухтияров,
заместитель директора ИОХ РАН, д.х.н. Александр Юрьевич Стахеев
В конференции приняли участие 200 специалистов из 30 городов 10 стран мира, в том числе:
из России – 181; Казахстана – 4, США – 4, Польши – 3, Испании – 3; Германии, Италии, Японии,
Нидерландов, Великобритании – по 1 представителю.

На переднем плане: один из первых учеников Г.К. Борескова,
директор Института углехимии и химического материаловедения СО РАН,
чл.-корр. РАН Зинфер Ришатович Исмагилов (второй слева) и делегация из Алматы, Казахстан
В течение трех рабочих дней участники конференции прослушали и обсудили 3 пленарные лекции, 10 ключевых лекций, 26 устных (20 мин), а также 92 стендовых доклада по следующим научным направлениям: 1) Формирование активного состояния катализатора под воздействием реакционной среды; 2) Формы кислорода на поверхности катализаторов: локальная структура, электронное строение и реакционная способность; 3) Структурная чувствительность, размерный эффект и катализ на одиночных атомах; 4) Механизмы окислительных реакций; 5) От кинетики реакции к промышленным инновационным процессам.
На церемонию открытия в Большом зале Дома ученых собрались участники конференции и сотрудники Института катализа СО РАН.

Открыл конференцию директор Института катализа академик РАН В.И. Бухтияров
памятной лекцией о жизни и научной деятельности Георгия Константиновича Борескова

Профессор Валерий Соколовский, Rennovia Inc., США
В первой пленарной лекции профессор Валерий Соколовский (Rennovia Inc., Санта Клара, США) рассказал о перспективах применения гетерогенных каталитических процессов для получения ценных химических соединений (диолы, диамины и органические кислоты) из возобновляемого органического сырья (крахмал, сахара, целлюлоза).
Им также были освещены проблемы разработки катализаторов, устойчивых к зауглероживанию и коррозии в растворах органических соединений, с использованием комбинаторных подходов к синтезу и испытанию катализаторов; продемонстрирована успешная реализация на пилотной уровне процессов получения гександиола из фруктозы и адипиновой кислоты из глюкозы с помощью разработанных катализаторов, в том числе на основе платины и других металлов, нанесенных на диоксид циркония.

Профессор Малгожата Витко (Malgorzata Witko), Институт катализа и химии поверхности, Краков, Польша
Профессор Малгожата Витко (Директор Института катализа и химии поверхности, Краков, Польша) в пленарной лекции обсудила проблемы активации кислорода на металл-оксидных системах (оксидах молибдена, вольфрама, гетерополикислот, металлопорфиринах) с использованием квантово-химического метода функционала плотности в нескольких вариантах. В лекции были проанализированы взаимосвязь локальной структуры поверхности и координации катионов с энергетикой удаления различных форм кислорода, определяющей их реакционную способность, а также взаимодействия молекулярного кислорода с образующимися вакансиями, в том числе с образованием его активированных форм (пероксидных и озонидных). Показана повышенная реакционная способность таких форм по отношению к взаимодействию с восстановителями (водород, оксид углерода).
Пленарный лектор профессор Ватару Уеда (Wataru Ueda, Университет Канагавы,Япония) представил результаты разработки новых микропористых оксидов на основе оксидов молибдена и вольфрама, исследования их структуры и текстуры и путей активации кислорода. Показано, что такие катализаторы отличаются уникальными каталитическими свойствами в реакциях селективного окисления этана и альдегидов, а также в окислении метакролеина в метакриловую кислоту.
Важным событием конференции стал Круглый стол «Идеи Г.К. Борескова и современность». Идеи и принципы Г.К. Борескова о системном подходе к катализу – от научных основ предвидения каталитического действия, приготовления катализаторов до расчета контактных аппаратов и промышленной реализации каталитических процессов – в наше время стали международным стандартом.

С теплыми воспоминаниями о совместной научной и научно-организационной работе
с Георгием Константиновичем выступил научный руководитель ИПХЭТ СО РАН
академик РАН Геннадий Викторович Сакович
С большим интересом были заслушаны сообщения директора Института проблем переработки углеводородов СО РАН к.х.н. Александра Валентиновича Лавренова и директора по развитию АО «СКТБ «Катализатор» Сергея Петровича Кильдяшева – идея создания этих организаций принадлежала Георгию Константиновичу Борескову.

Участники Круглого стола «Идеи Г.К. Борескова и современность»
Генеральный директор ЗАО «Самарский завод катализаторов» Антон Андреевич Копытин обратил внимание участников на усовершенствованные высокоэффективные сернокислотные ванадиевые катализаторы на силикагелевых и диатомитовых носителях, которые завод выпускает по лицензии Института катализа с 2002 года. История создания барий-алюмо-ванадиевого катализатора уходит 1930-е годы – катализатор был разработан и освоен в промышленности под руководством Г.К. Борескова. Выступление представителя Фонда инфраструктурных и образовательных программ (ООО «УК «РОСНАНО») к.х.н. Наталии Александровны Емашовой позволило аудитории ознакомиться с деятельностью фонда в области популяризации нанотехнологий и наноиндустрии.
Содержательные секционные ключевые лекции были представлены известными учеными.

Профессор Грэм Хатчинс (Graham Hutchings Университет Кардиффа, Великобритания)
читает ключевую лекцию
В ключевой лекции профессора Грэма Хатчинса (Graham Hutchings, Университет Кардиффа, Великобритания) были рассмотрены проблемы создания нанокомпозитных катализаторов на основе наночастиц сплавов золота на оксидных носителях. Показана высокая активность и стабильность таких катализаторов в реакциях прямого синтеза перекиси водорода путем гидрирования молекулярного кислорода и селективного окисления спиртов и углеводородов.
В ключевой лекции профессора Фабрицио Кавани (Fabrizio Cavani, Университет Болоньи, Италия) «Катализаторы получения синтетического био-бутадиена: от промышленного применения к механизму реакции» рассмотрены проблемы получения данного соединения путем одностадийной трансформации биоэтанола в рамках процесса Лебедева, разработанного в России в 30-е годы. Применение методов ИКС in situ показало, что механизм образования бутадиена включает стадию прямого взаимодействия ацетальдегида и этанола с образованием кротилового спирта, который затем дегидратируется с образованием бутадиена. Теоретический анализ с помощью метода функционала плотности позволил установить атомно-молекулярные причины, обеспечивающие самую высокую активность и селективность в данной реакции катализатора MgO/SiO2. Промотирование этого катализатора Ga3+ позволяет улучшить его каталитические свойства.
В ключевой лекции профессора Эмиля Хенсена (Emiel J.M. Hensen, Технический университет Эндховена, Голландия) «Дегидроароматизация метана – новые взгляды и возможности» рассмотрены проблемы повышения устойчивости к закоксовыванию систем Mo-ZSM-5, используемых в промышленности в качестве катализаторов данной реакции. Показано, что это может быть достигнуто периодическим введением импульсов кислорода в поток реакционной смеси, что позволяет также получить синтез-газ в качестве ценного побочного продукта. Другой подход основан на снижении содержания молибдена в катализаторе, что позволяет использовать синтетический воздух для регенерации катализатора при температуре реакции.
В ключевой лекции профессора Жозе Конесы (José Carlos Conesa, Институт катализа и нефтехимии, Мадрид, Испания) «Исследование катализатора селективного окисления СО CuOx/CeO2 с использованием методов операндо на синхротронном излучении и функционала плотности» рассмотрена проблема регулирования свойств активных центров такого катализатора, расположенных на границе раздела металл-оксид. Показано, что использование носителя – нанокубического CeO2 с развитыми рыхлыми гранями (001) позволяет получить более высокую активность и селективность окисления примеси СО в потоке водорода по сравнению с микросферическим носителем, что объяснено более сильным взаимодействием меди с носителем, приводящим к стабилизации ее катионных форм.
В ключевой лекции профессора Ангелики Брюкнер (Angelika Brückner, Институт катализа им. Лейбница, Росток, Германия) «Что вы видите – это (не) то, что у вас есть, почему и нужна операндо спектроскопия для установления природы активных центров, получаемых из предшественников» данная проблема рассмотрена на примере двух реакций – окислительной димеризации бутена на нанесенных никелевых катализаторах Ni/SiO2-Al2O3 и электрокаталитического выделения кислорода на смешанных CoNi и CoCu оксидных катализаторах с использованием методов XANES/EXAFS, ЭПР и комбинационного рассеяния в реакционных условиях. Показано, что для первой реакции доля активных центров – катионов Ni+ существенно уменьшается по сравнению с исходным состоянием. Для второй реакции в щелочных растворах на поверхности катализаторов происходит образование разупорядоченного слоя оксо-гидроксидов никеля и кобальта, что приводит к росту активности.
В ключевой лекции академика РАН Валентина Николаевича Пармона (Институт катализа им. Г.К. Борескова СО РАН) «Катализ и энергетика: опыт Института катализа» рассмотрены проблемы разработки катализаторов и технологий для получения высококачественного топлива из традиционных углеводородов, переработки попутного газа и энергетики будущего. Последний аспект включает каталитическое сжигание различного вида топлив, переработку возобновляемого растительного сырья, термокаталитические процессы для преобразования ядерной и солнечной энергии в химическую, а также рекуперации низкопотенциального тепла с использованием композитных адсорбентов воды.
В ключевой лекции профессора Марка Вениаминовича Цодикова (Институт нефтехимического синтеза РАН, Москва) «Перспективные каталитические реакции производства углеводородных компонентов топлив и C4-C8 спиртов из биотоплив» были рассмотрены процессы трансформации этанола и других спиртов, смеси органических продуктов ферментации, глицерина и рапсового масла в алканы, олефины, ароматические соединения, нафтены и кислородсодержащие компоненты газолина и дизельного топлива. В этих процессах использовались как коммерческие, так и новые катализаторы на основе наночастиц биметаллических активных компонентов I, II, V-VIII групп, нанесенных на оксид алюминия или цеолиты. Структура и природа активных центров катализаторов были исследованы с помощью температурно-программированной десорбции молекул-тестов, ЕXAFS, РФЭС, рентгеноструктурного анализа и электронной микроскопии высокого разрешения, что позволило установить из взаимосвязь с каталитическими свойствами. Показана перспективность ряда катализаторов и процессов для практического применения.

Ключевая лекция профессора Марка Вениаминовича Цодикова (ИНХС РАН, Москва)
Чл.-корр. РАН Зинфер Ришатович Исмагилов в своей лекции «Окислительная десульфуризация дизельных фракций: от исследований механизма к перспективной промышленной технологии» (Институт углехимии и химического материаловедения СО РАН, Кемерово) рассказал о проблемах удаления алкил-замещенных дибензотиофенов путем газофазного окисления. Показано, что катализаторы на основе CuZnAl-O, модифицированного оксидами висмута и молибдена, обладают необходимой активностью, селективностью и стабильностью в данной реакции.

Ключевая лекция доктора Джеймса Рекоске (James Rekoske, ЮОП-Хонивелл, США)
В ключевой лекции доктор Джеймс Рекоске (James Rekoske, ЮОП-Хонивелл, США) «Последние достижения в нефтехимических технологиях получения олефинов и ароматики» рассмотрел проблемы разработки эффективных наноструктурированных катализаторов дегидрирования алканов и транс-алкилирования ароматики с получением параксилола. Для первого процесса были созданы наноструктурированные полиметаллические катализаторы, обладающие уникально высокой активностью, селективностью и стабильностью, обеспечивающей высокую экономическую эффективность. Для второго процесса предложен новый эффективный катализатор на основе синтетического морденита с наноразмерными частицами, превосходящий по своим свойствам коммерческий катализатор.
В ключевой исторической лекции профессора Михаила Борисовича Розенкевича (Российский химико-технологический университет имени Д.И. Менделеева, Москва) «Основанный Г.К. Боресковым отдел в Университете имени Д.И. Менделеева – от 1949 года до наших дней» был представлен обзор деятельности отдела технологии разделения и применения изотопов легких химических элементов. Во время работы в качестве начальника отдела до 1960 года Г.К. Боресков создал новое направление исследований «Изотопы в катализе», включающее как применение изотопных методов в исследовании катализаторов, так и разработку катализаторов для разделения стабильных изотопов, которое успешно развивается и в настоящее время. В практическом плане наиболее важным достижением отдела явилась разработка катализаторов для орто-пара конверсии водорода и разделения изотопов водорода путем химического обмена водорода и воды, за которую сотрудники отдела Р.А. Буянов и М.Г. Слинько получили Ленинскую премию 1960 года. В фундаментальном плане наиболее значимыми были исследования влияния эффектов наноструктуры катализаторов на основе редкоземельных металлов и металлов платиновой группы в реакциях орто-пара конверсии водорода и гомомолекулярного изотопного обмена.
Устные доклады были посвящены в основном изучению новых катализаторов на основе нанесенных наночастиц металлов.Так, доклад к.х.н. Д.А. Булушева (Институт катализа СО РАН, Новосибирск) был посвящен изучению катализаторов на основе платины для производства водорода. Используя современные методы, автор убедительно продемонстрировал высокую каталитическую активность кластеров платины в реакции разложения муравьиной кислоты. Профессор Кортес Корберан (Vicente Cortes Corberan, Институт катализа, Мадрид, Испания) рассказал об исследовании реакции окисления октанола на золотых катализаторах. Тему активности наночастиц золота в каталитических гетерогенных реакциях продолжила д.х.н. Д.А. Пичугина (МГУ, Москва), которая представила результаты квантово-химических расчетов адсорбционных свойств нанесенных наночастиц золота. Доклад доктора Дороты Рутковска-Збик (Dorota Rutkowska-Zbik, Институт катализа и химии поверхности, Краков, Польша) был посвящен изучению реакции гидрирования CO2 в метанол. Было показано, что в данной реакции можно успешно применять катализаторы на основе оксида циркония. Доктор Роберт Косидар (Robert Kosydar) из того же института представил результаты исследования реакции гидрирования ненасыщенных альдегидов на поверхности наночастиц палладия. В выступлении аспиранта Института катализа СО РАН А.К. Худорожкова были рассмотрены результаты исследований размерного эффекта в реакции окисления пропана на палладии.
К.х.н. И.В. Мишаков (Институт катализа СО РАН) посвятил свой доклад изучению стабильности катализаторов на основе никеля. К.х.н. И.Г. Данилова (Институт катализа СО РАН) сообщила об исследовании катализаторов на основе нанесенных наночастиц палладия в реакции окисления СО. К.х.н. Е.А. Козлова, также из Института катализа СО РАН, обсудила результаты исследования фотокатализаторов на основе меди и сульфидов кадмия и цинка в процессе получения водорода. Д.х.н. К.П. Волчо (НГУ, Новосибирск) рассказал об уникальных каталитических свойствах наночастиц золота. О.Г. Сальников (Международный томографический центр СО РАН, Новосибирск) сообщил об исследовании катализаторов на основе металлов платиновой группы с использованием метода ядерного магнитного резонанса. Доклад к.х.н. Н.В. Максимчук (Институт катализа СО РАН) был посвящен жидкофазному окислению субстратов на ниобий-содержащих катализаторах.
Профессор В.А. Садыков (Институт катализа СО РАН) рассказал об исследовании окислительно-восстановительных реакций на оксидных катализаторах. К.х.н. Е.В. Староконь (Институт катализа СО РАН) представил последние результаты по исследованию механизма эпоксидирования легких алканов кислородом на поверхности катализатора FeZSM-5. В докладе к.х.н. А.В. Матвеева (Институт катализа СО РАН) были представлены данные по квантово-химическому моделированию реакционной способности поверхностного кислорода в окислении угарного газа на металлическом палладии. К.х.н. С.А. Яшник (Институт катализа СО РАН) осветила актуальную тематику исследования катализаторов окисления метана. Доктор Алак Батачария (Alak Bhattacharyya, Honeywell UOP, США) рассказал об опыте сотрудников компании в разработке катализаторов для промышленных приложений. Д.х.н. Б.Н. Кузнецов (ФИЦ «Красноярский научный центр СО РАН») осветил вопросы переработки древесной биомассы. Д.х.н. С.Ф. Тихов (Институт катализа СО РАН) рассказал об изучении механизма окисления угарного газа на цериевых катализаторах. Д.х.н. Т.П. Минюкова (Институт катализа СО РАН) доложила о проблемах контроля реакционной способности наночастиц меди, нанесенных на различные носители, в превращениях синтез-газа. Д.х.н. А.С. Белый (ИППУ СО РАН, Омск) рассказал о текущем состоянии и перспективах развития процесса каталитического риформинга.
Д.х.н. М.М. Слинько (Институт химической физики РАН, Москва) рассказала о последних результатах в области изучения нелинейных явлений в гетерогенных каталитических системах. В докладе к.ф.-м.н. Е.А. Лашиной (Институт катализа СО РАН) были затронуты вопросы исследования механизма осцилляций в парциальном окислении метана на металлическом никеле. Большое внимание привлек доклад д.х.н. А.И. Боронина (Институт катализа СО РАН) о современной интерпретации концепций, предложенных Г.К. Боресковым, на примере окисления монооксида углерода. К.х.н. Д.И. Потемкин (Институт катализа СО РАН) рассказал о последних результатах в области изучения модельных катализаторов избирательного окисления угарного газа. В заключение сессии устных докладов Конференции выступил к.х.н. А.Р. Чолач (Институт катализа СО РАН) с докладом, посвященным изучению активных центров катализаторов синтеза аммиака.
В рамках молодежной программы конференции cостоялась Школа-симпозиум молодых ученых «In situ и Operando исследования каталитических реакций». В программу были включены две ключевые лекции академика РАН В.И. Бухтиярова (Институт катализа СО РАН) и профессора Мигеля Баньяреса (Miguel A. Bañares, Институт катализа, Мадрид, Испания). Лекция В.И. Бухтиярова была посвящена изучению формирования активных центров катализаторов с помощью in situ методов. На примере реакций эпоксидирования этилена и окисления углеводородов было продемонстрировано, как реакционная среда влияет на фазовый и химический состав активного компонента. Лекция М.А. Баньяреса была посвящена применению метода Рамановской спектроскопии в режиме in situ для исследования нанесенных катализаторов. На примере реакций окисления парафинов до олефинов и нитрилов были показаны возможности in situ Рамановской и ИК-спектроскопии.
На Школе были также представлены девять устных докладов молодых ученых. К.х.н. А.А. Габриенко из Института катализа СО РАН представил результаты исследования активного компонента катализатора H-ZSM-5 в реакции окисления метана. С помощью метода in situ твердотельной ЯМР спектроскопии был изучен процесс активации метана на Zn2+ центрах. Возможности применения in situ ЯМР спектроскопии для определения интермедиатов реакции продемонстрировала Д.Б. Буруева из Международного томографического центра СО РАН, Новосибирск. В докладе к.ф.-м.н. С.С. Якушкина (Институт катализа СО РАН) были представлены результаты in situ ЭПР исследований металл-оксидных интерфейсов в катализаторе Au/Al2O3, что в дальнейшем позволило установить механизм гидрирования нитробензола на данных катализаторах. Тему применения in situ ЭПР продолжил доклад к.х.н. О.Ю. Лякина из Института катализа СО РАН. Его работа была посвящена исследованию энантиоселективности в реакциях эпоксидирования на биомимитических железных катализаторах. Доклад З.С. Винокурова из Института катализа СО РАН был посвящен исследованию автоколебаний при окислении метана на палладиевом катализаторе. Применение метода in situ РФА позволило установить определить периодическое окисление-восстановление палладия, а также наличие фазы PdCx. В докладе к.х.н. Д.В. Красникова (Институт катализа СО РАН) были представлены результаты исследования активации биметаллических катализаторов формирования многостенных углеродных нанотрубок с помощью метода in situ РФА. Доклад к.х.н. О.А. Булавченко (Институт катализа СО РАН) был посвящен исследованию процесса восстановления смешанного Mn-Zr катализатора окисления CO. С помощью методов in situ РФЭС и РФА было показано, что восстановление марганца происходит в несколько этапов, при этом часть марганца сегрегируется на поверхности оксида. В докладе А.Н. Субоч (Институт катализа СО РАН) были представлены результаты исследования синтеза азот-допированных углеродных нанотрубок, которые являются перспективными катализаторами селективного окисления алкиларенов. В заключительном докладе Школы Э.И. Карычевой (Институт катализа СО РАН) было доложено об in situ исследованиях процесса агрегации оксида титана в органической среде.
Организационный комитет наградил почетными Дипломами и денежными премиями двух молодых участников Школы-симпозиума, продемонстрировавших высокие достижения в применении in situ методов в исследованиях гетерогенных каталитических реакций.

К.х.н. О.А. Булавченко была удостоена Диплома Российского химического общества им. Д.И. Менделеева
за лучшую работу в области использования методов рентгеновской дифракции и рентгеновской фотоэлектронной спектроскопии для in situ исследований оксидных катализаторов окисления СО
К.х.н. Д.В. Красников получил почетный Диплом Конференции за успехи в in situ исследованиях активации биметаллических катализаторов синтеза многостеночных углеродных нанотрубок.
Для аспирантов Института катализа во время конференции прошел конкурс на получение стипендий академика Г.К. Борескова. Именная стипендия академика Г.К. Борескова была присуждена аспиранту И.В. Шаманаеву для продолжения изучения закономерностей превращения органических кислородсодержащих соединений в присутствии катализаторов на основе фосфидов переходных металлов.


Стендовая сессия собрала большую и активную аудиторию:
профессор Кортес Корберан, Институт катализа, Мадрид, и Ирина Красникова, Институт катализа, Новосибирск (фото 1);
д.х.н. Александр Викторович Хасин и д.х.н. Тамара Витальевна Андрушкевич,
Институт катализа, Новосибирск (фото 2)
Брошюра с Научной программой конференции и сборник тезисов на флеш-картах были предоставлены участникам при регистрации. Электронному изданию сборника тезисов присвоен Международный стандартный книжный номер (ISBN-978-5-906376-16-9).
Прошедшая Конференция позволила провести анализ полученных результатов и обсудить перспективные направления исследований в фундаментальных и прикладных направлениях развития гетерогенного окислительного катализа – основной области интересов научной школы академика Г.К. Борескова.

Успех многих работ, представленных на Конференции, был связан с применением современных физических методов для исследования состояния катализаторов в условиях протекания каталитических реакций, а также теоретических методов состояния и реакционной способности кислорода на их поверхности. В области синтеза новых катализаторов были продемонстрированы хорошие результаты в дизайне нанокомпозитных и наноструктурированных катализаторов, в том числе на основе наночастиц металлов и сплавов, нанесенных на оксидные носители, обладающих высокой эффективностью и стабильностью в широком круге промышленно важных реакций, включая реакции трансформации традиционных углеводородов и биомассы в ценные химические продукты, процессы водородной энергетики, сероочистки топлив, термокаталитические процессы для преобразования ядерной и солнечной энергии в химическую, а также процессы рекуперации низкопотенциального тепла с использованием композитных адсорбентов воды.
На церемонии закрытия Конференции были подведены итоги и дана высокая оценка работы Организационного комитета и Совета научной молодежи Института катализа по подготовке и проведению международного мероприятия.

Диплом Официального спонсора вручается директору Института проблем переработки
углеводородов СО РАН к.х.н. Александру Валентиновичу Лавренову
Было почеркнуто, что молодые ученые умело продемонстрировали результаты своих исследований в докладах на Конференции и Школе.

Одна из первых учениц Г.К. Борескова, доктор химических наук Тамара Витальевна Андрушкевич,
автор книги о Георгии Константиновиче из серии «Материалы к библиографии ученых СССР»
В продолжение и сохранение традиции предыдущих конференций памяти академика Г.К. Борескова 1987 – 1997 – 2007 – 2017 годов, а также в целях повышения квалификации научной молодежи, было решено провести V Конференцию БОРЕСКОВСКИЕ ЧТЕНИЯ в 2027 году.
Материал подготовили В.И. Бухтияров, А.А. Ведягин, В.В. Каичев, В.А. Садыков,
Е.А. Козлова, Л.Я. Старцева, М.С. Суворова (ИК СО РАН, Новосибирск)
Фото: А.А. Спиридонов, А.М. Ершова
Конференция памяти академика Борескова стала прекрасным поводом поговорить в кулуарах с ее участниками о Георгии Константиновиче.
Один из крупнейших мировых специалистов в области катализа профессор Грэм Хатчингс, директор Института катализа Университета Кардиффа, Великобритания, выступивший на конференции с ключевой лекцией «Катализ с использованием новых наноматериалов», рассказал следующее: «Я встречался с Боресковым, когда работал в ICI (Imperial Chemical Industries – британская химическая компания, считалась крупнейшей производственной компанией в Британской империи, вошла в 2007 году в состав AkzoNobel, – прим. ИК СО РАН). Боресков посещал наши лаборатории, когда я там работал, будучи очень молодым исследователем. Я, конечно, знал, что он был чрезвычайно важным человеком в области катализа, и знал о многих его работах, которые он написал в те дни. Многие его статьи были опубликованы в одном русском журнале, который был переведен на английский. Кажется, журнал назывался «Кинетика и катализ», я брал его в библиотеке (тогда у нас было несколько подобных русских журналов, которые переводились на английский). В нем печатали очень короткие статьи, но в них было много интересных идей. Я смог их использовать в проектах, над которыми работал в промышленности. Мы осознавали значимость этих идей и работали с ними. Вот в чем проявилось для меня влияние Борескова. После работы в промышленности, когда я перешел в науку в 1984 году, у меня появилось много идей, которые я хотел бы привнести в академические круги. Поэтому мои научные работы были в значительной степени подвержены влиянию всего того, что я делал в промышленности».

Профессор Грэм Хатчингс (Graham Hutchings) с супругой Салли Хатчингс (Sally Hutchings) в Доме ученых СО РАН
Говоря о конференции, профессор Хатчингс снова сделал ударение на важности связи науки и промышленности и, в частности, похвалил ключевую лекцию доктора Джеймса Рекоске из компании Honeywell UOP, США (Honeywell UOP – многонациональная компания, разрабатывающая и поставляющая технологии для нефтеперерабатывающей, газоперерабатывающей, нефтехимической и крупной обрабатывающей промышленности, – прим. ИК СО РАН): «Вы привлекли чрезвычайно интересных людей, как на местном, так и на международном уровне. Презентация UOP была отличной. Это фантастика - иметь такие связи с промышленными компаниями, как у Института катализа им. Борескова, и работать вместе как одна команда. Очень немногие конференции имеют реальную связь с промышленниками. Это то, что нужно поощрять и развивать».
Профессор Хатчингс также выделил активное участие молодежи в работе конференции: «Молодые исследователи полны энтузиазма. Они очень увлечены, хотят задавать вопросы, хотят участвовать в дебатах, и это действительно интересно. Стендовая сессия была исключительно живой – обсуждалась настоящая наука».
Еще один участник ключевой сессии докладов, заведующий лабораторией Института нефтехимического синтеза им. А.В. Топчиева РАН, доктор химических наук, профессор Марк Вениаминович Цодиков тепло отозвался о Георгии Константиновиче Борескове и об Институте катализа, создание которого Боресков считал основным делом своей жизни: «Академик Боресков, наверное, для меня символ. Я его застал один раз и сейчас горжусь этим. Георгий Константинович был основоположником всего сернокислотного производства. Это известно всей стране и всему миру. Поэтому он вошёл в число великих химиков-каталитиков. С другой стороны, всё-таки я «каталитический человек» и старый друг Института катализа. Поэтому у меня очень большое уважение к Георгию Константиновичу за то, что он создал такой хороший институт. У меня здесь много товарищей, которые очень много мне помогали в моих работах в свое время. Некоторых уже нет в живых. Они дали мне очень многое, может быть, наибольшее из того что есть. Мне всегда нравится, как здесь устраиваются конференции, потому что коллеги очень серьезно к этому относятся, всех участников захватывает наука, атмосфера конференций всегда вдохновляет. Интересно приехать и проверить себя, насколько ты преуспел, пообщаться с коллегами, зарядиться. Приезжаешь и погружаешься в работу, ощущая полное взаимопонимание коллег. Это все очень здорово».

Доктор химических наук Марк Вениаминович Цодиков и кандидат
химических наук
Лариса Александровна Аркатова, АО «СКТБ «Катализатор»
Профессор Валерий Соколовский (Rennovia Inc., США) – один из любимых учеников Георгия Константиновича – рассказал о своем учителе: «Начну с того, что вообще в катализе я оказался благодаря моей встрече с Боресковым. Моя специальность после университета была «разделение изотопов», чем я очень гордился. В то время все, что касалось атома, было очень популярно. Но однажды мой руководитель дипломной работы сказал мне: «Я собираюсь на встречу с Боресковым. Он организует Институт катализа Академии наук в Сибири. Сейчас он в Москве. Хочешь, пойдем со мной, думаю, тебе будет интересно». Я подумал: «Академия наук интересно, но причем тут катализ, если я собираюсь заниматься изотопами. Тем не менее, я решил пойти. Боресков произвел на меня такое впечатление, что я подумал: такая возможность предоставляется раз в жизни и тут же согласился ехать в Сибирь к нему в Институт. Мне посчастливилось проработать с ним более 20 лет. Конечно, я многое перенял у него в моем отношении к науке, да и к жизни вообще. Это был необыкновенный человек. Можно сказать, что он был умным и талантливым. Но сказать это – ничего не сказать. Я встречал в своей жизни очень умных и талантливых людей, но никого из них я не могу поставить рядом с Боресковым».

Слева направо: профессор Геннадий Иванович Панов,
профессор
Валерий Соколовский и профессор Владимир Александрович
Собянин
Говоря о конференции, профессор Соколовский отметил важность того, что «Боресковские чтения позволяют новому поколению исследователей приобщиться к тем идеям, которые были заложены при организации Института катализа и которые не потеряли свою ценность спустя много лет».
Для завершения описания юбилейной конференции прекрасно подойдут высказывания самого Георгия Константиновича Борескова, которые привел его ученик профессор Соколовский:

Материал подготовила А.М. Ершова (ИК СО РАН, Новосибирск)
 20 апреля 2017 года исполнилось 110 лет со дня рождения академика Георгия Константиновича Борескова –
выдающегося русского ученого в области физической химии, катализа и химической инженерии, замечательного педагога и уникального
руководителя.
20 апреля 2017 года исполнилось 110 лет со дня рождения академика Георгия Константиновича Борескова –
выдающегося русского ученого в области физической химии, катализа и химической инженерии, замечательного педагога и уникального
руководителя.
Вехи биографии
Георгий Константинович родился в 1907 году в Омске. В 1929 году окончил Одесский химический институт. Научная деятельность ученого началась в том же году в Одесском химико-радиологическом институте в лаборатории катализа с разработки катализатора для контактного производства серной кислоты. Разработанный Георгием Константиновичем ванадиевый катализатор имел исключительную важность и применялся во время Великой Отечественной войны на заводах страны для получения серной кислоты – необходимого компонента взрывчатых веществ.
Работы в области катализа Г.К. Боресков продолжил в Москве, куда была переведена его лаборатория, сначала в Научно-исследовательском институте удобрений и инсектофунгицидов (с 1937 по 1946 гг.), а затем в Научно-исследовательском физико-химическом институте им. П.Я. Карпова (с 1946 по 1959 гг.).
В только что возникший Новосибирский Академгородок Г.К. Боресков приехал в 1961 году уже будучи крупным ученым с мировым именем. Здесь он организовал Институт катализа СО АН СССР и оставался его бессменным руководителем на протяжении 26 лет (с 1958 по 1984 гг.). В 1992 году Институту катализа СО РАН было присвоено имя Георгия Константиновича Борескова.

Завершение строительства здания Института, май 1963 г.
Создание Института катализа Боресков считал основным делом своей жизни. Он был лидером всех научных направлений Института: от научных основ прогнозирования каталитического действия и приготовления катализаторов до промышленной реализации каталитических процессов. Комплексный подход к изучению катализа в наше время стал международным стандартом.
Научный вклад
Научный вклад Георгия Константиновича Борескова трудно переоценить. В своей деятельности он придерживался лучших традиций классиков русской химии – Д.И. Менделеева, А.Е. Фаворского, Н.Д. Зелинского, – объединяя глубокие фундаментальные исследования, которым он придавал исключительно важное значение, с решением насущных практических задач.
Г.К. Боресков являлся убежденным сторонником химического подхода к катализу, согласно которому механизм каталитического действия заключается в промежуточном химическом взаимодействии катализатора с реагирующими веществами. Его концепция катализа как исключительно химического явления имела особое значение для создания современных физико-химических основ катализа.

Заседание
Президиума СО АН СССР в доме М.А. Лаврентьева, 1961 г.
Слева
направо: Г.К. Боресков, М.А. Лаврентьев, Н.Н. Ворожцов, С.Т. Беляев
Одним из главных принципов, вытекающих из химического подхода к катализу, является взаимосвязь и взаимозависимость катализатора и реагирующих веществ: каждому составу реакционной смеси и температуре отвечает определённое состояние катализатора, отличающееся от его исходного состояния. Катализаторы являются лабильными компонентами реакционной системы и под воздействием реакционной смеси меняют химический состав, структуру поверхности и каталитические свойства. При одинаковом химическом составе и структуре катализатора каталитическая активность, отнесённая к единице величины поверхности (удельная каталитическая активность, УКА), приблизительно постоянна. УКА (specific surface activity) принята мировым сообществом как мера активности катализатора, а принцип постоянства УКА вошел в науку о катализе под названием "правило Борескова".
Необходимость всестороннего изучения системы «катализатор-реакция» Георгий Константинович особо подчёркивал при подборе катализаторов и создании кинетической модели гетерогенной каталитической реакции.
Работы Борескова в области кинетики обратимых реакций имеют основополагающее значение для теории кинетики сложных реакции. Он установил общее соотношение между энергиями активации прямой и обратной реакции с учётом молекулярности реакции и предложил методы её определения.
Георгий Константинович Боресков первым стал широко применять химическую кинетику в инженерных расчётах. Из его ранних работ по сернокислотному производству возникло новое направление – математическое моделирование химических процессов, которое сейчас является теоретической основой химической технологии.
Ученый. Учитель. Человек
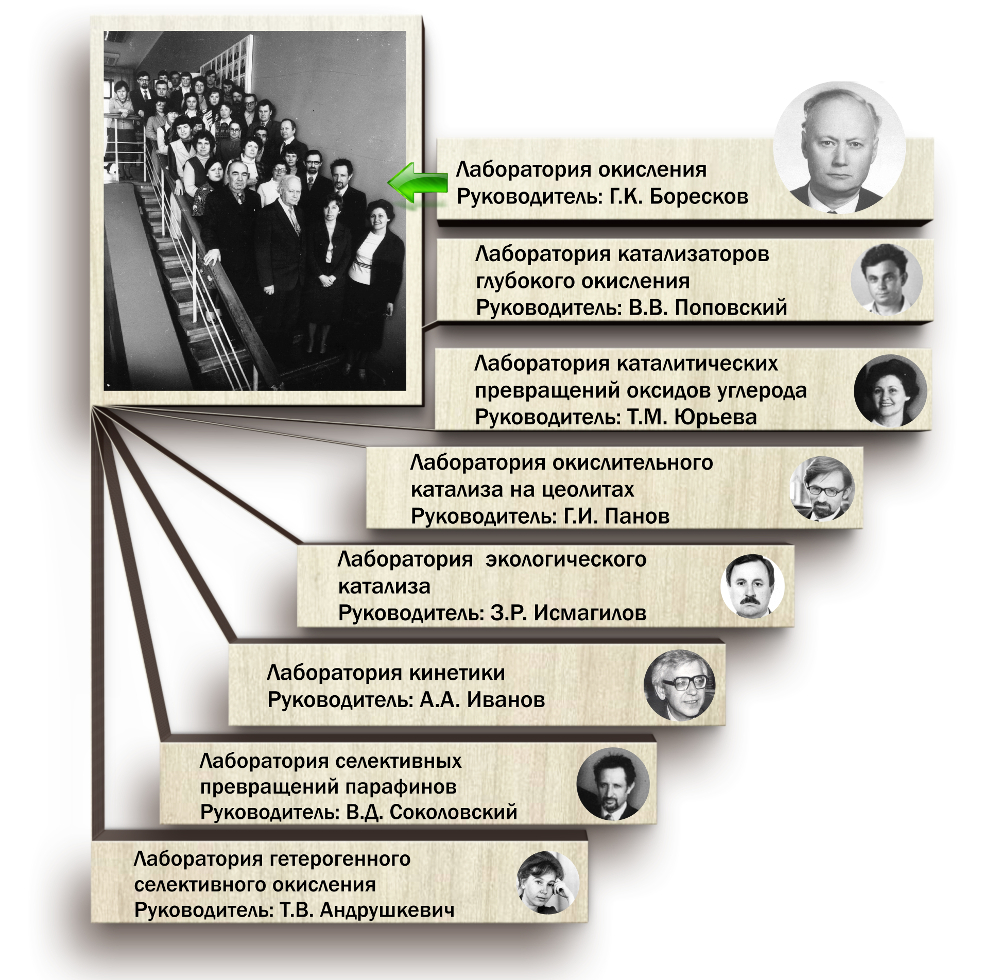
Лаборатории, «выросшие» из Лаборатории
окисления, которой руководил Г.К. Боресков,
а также их первые
руководители – ученики Георгия Константиновича
Георгий Константинович много времени уделял педагогической работе, которую начал одновременно с научными исследованиями и всегда неразрывно связывал с ними. Около 11 лет он руководил кафедрой разделения и применения изотопов в Московском химико-технологическом институте им. Д.И. Менделеева, а позднее – основанной им в Новосибирском государственном университете кафедрой катализа и адсорбции. Георгий Константинович возглавлял лабораторию окисления и создал научную школу, в которой выросли известные специалисты, плодотворно работающие в области катализа, чьи исследования подтверждают и развивают идеи Борескова.
Творческое наследие Г. К. Борескова составляет более 800 статей. Он является автором монографий "Технологии серной кислоты", "Технология процессов химического изотопного обмена", "Гетерогенный катализ".
Георгий Константинович обладал огромным личным магнетизмом, глубокими знаниями, богатейшим опытом, неиссякаемым энтузиазмом и всегда создавал вокруг себя уникальную творческую среду. Его отличительными качествами были верность принципам, требовательность к себе, работоспособность и самодисциплина в сочетании с доброжелательностью, интеллигентностью и терпимостью. Аристократ по происхождению и по духу, выдающийся специалист, он стал для своих учеников примером ученого и человека.
Членство в организациях
| Член Президиума СО АН СССР |  |
| Представитель СССР в Совете уполномоченных стран СЭВ по проблеме «Промышленные катализаторы» | |
| Председатель проблемной комиссии «Кинетика и катализ» Академий наук социалистических стран | |
| Почетный член Нью-Йоркской академии наук | |
| Иностранный член Германской академии наук | |
| Почетный доктор Вроцлавского политехнического института | |
| Почетный доктор университета в г. Пуатье |
Государственные награды
Лауреат Сталинской премии III (1942) и II (1953) степени
Лауреат Государственной премии Украинской ССР (1970)
Герой Социалистического Труда (1967)
Награжден орденами Ленина (1967, 1975, 1982), Трудового Красного Знамени (1952), наградой «Знак Почета» (1944)
Сайт Института катализа им. Г.К. Борескова СО РАН
Approach greatly improves customizability and efficiency of poly(α-hydroxyacid) synthesis

A two-catalytic-step process can efficiently convert versatile OCA monomers into a range of customizable PAHAs.
Poly(α-hydroxy acid), or PAHA, polyesters can form products such as biodegradable sutures and implants, as well as cell-penetrating nanoparticles for drug and gene delivery. But synthesizing PAHA polyesters hasn’t been easy. Now researchers report a new way to prepare PAHA polyesters with desirable sets of properties that have been difficult or impossible to access before.
The stiffness, stretchability, tensile strength, and other physical properties of these polymers can be customized by varying the sidechains of the lactide and glycolide monomers used to make the materials. The repertoire of PAHA polyesters has been limited because synthesizing these monomers requires multistep and low-yield reactions, and adding sidechains to them is difficult.
In 2006, Didier Bourissou of Paul Sabatier University and coworkers showed that PAHAs could instead be made from O-carboxyanhydride (OCA) monomers, which are much easier to prepare and modify. Nevertheless, the organocatalytic reactions used to polymerize OCAs are slow and have undesired side reactions. Also some of the polymer products have uncontrolled stereochemistry and broad and unpredictable molecular weight distributions.
Rong Tong and Quanyou Feng at Virginia Tech have now addressed these issues with a new method for rapid and controlled OCA polymerization (J. Am. Chem. Soc. 2017, DOI: 10.1021/jacs.7b01462). In the two-step approach, a photoredox nickel-iridium catalyst first decarboxylates OCAs. Then zinc alkoxide catalyzes a ring-opening polymerization of the decarboxylated OCAs to yield the PAHAs. The process is fast, minimizes undesired side reactions, and produces PAHAs with controlled stereochemistry and narrow and predictable molecular weight distributions.
“Our chemistry will allow us to prepare polyester materials with customized macroscopic properties such as rigidity, elasticity, and biodegradability,” Tong says. Virginia Tech has applied for a patent on the technique.
Bourissou agrees that the technique should make “a variety of PAHA polymers with different functionalities and microstructures readily accessible.” He points out that “one practical limitation is that the ring-opening polymerization has to be performed at low temperature, –15 °C or below, to achieve good control and avoid side-reactions.” Fortunately, he says, OCAs are highly reactive and polymerize rapidly even at low temperature.
A win-win approach to curbing climate change could be capturing carbon dioxide from the atmosphere and converting it to valuable products. A study presented at the American Chemical Society national meeting in San Francisco may help advance that effort by revealing mechanistic details of a catalytic process that converts CO2 to a commodity chemical—methanol.
On industrial scales, catalysts composed of copper and zinc oxide supported on alumina hydrogenate carbon monoxide and CO2 to methanol. But these catalysts have shortcomings, according to Ping Liu, a chemist at Brookhaven National Laboratory.
Speaking at a symposium sponsored by the Division of Energy and Fuels, Liu pointed out that the Cu-ZnO catalysts are not very efficient or selective in producing methanol. These reactions also require high temperatures and high pressures of the reactant gases. What’s more, she said, chemical details of the active catalytic site remain elusive. That information could be the key to designing catalysts with improved energy and chemical efficiency, Liu says.
In an ongoing debate regarding the catalyst’s active site, various researchers have argued that highly active Zn-Cu alloy species are the key catalytic players. In contrast, Liu’s new work suggests that the action occurs at the atomic interface between ZnO and Cu (Science 2017, DOI: 10.1126/science.aal3573).
To reach that conclusion, Liu, Brookhaven colleagues José A. Rodriguez and Shyam Kattel, and Columbia University’s Jingguang Chen, prepared several types of Cu and ZnO reference catalysts, including one made of zinc nanoparticles deposited on copper, and another with ZnO nanoparticles on copper.
They analyzed and directly compared the CO2-to-methanol chemistry of all the catalysts using synchrotron-based photoelectron spectroscopy and computational methods. The computations predicted that Cu-ZnO surface species should be the most reactive form of the catalyst. They also predicted that the Zn-Cu species shouldn’t remain stable under reaction conditions. Instead, it should react with oxygen and form copper zinc oxide. And that’s exactly what Liu and coworkers found in the lab.
Now the group aims to use that information to optimize the interface between ZnO and Cu to improve the catalysts.
“This is a highly important study with excellent quality data and supporting theoretical calculations,” said Charles T. Campbell, a catalysis specialist at the University of Washington, Seattle. CO2 hydrogenation to methanol is one of the most likely pathways for converting the greenhouse gas to a valuable product, Campbell asserted. He added that this study should help improve that catalytic process.
A team of chemists from industry and academia has taken a step back from the normal hustle and bustle of synthesizing new molecules to investigate what happens to workhorse palladium catalysts when they go catalyzing. The traditional thinking has been that a precursor Pd(II) salt is reduced to a catalytically active Pd(0) species when a ligand is introduced. Yet chemists have never had a complete picture of this reduction mechanism, and some scientists have found that under certain conditions a Pd(I) species can get involved in the formation of the Pd(0) active catalyst.
Researchers led by Franziska Schoenebeck, a chemistry professor at RWTH Aachen University, and Thomas J. Colacot, global R&D manager at catalyst firm Johnson Matthey, have now completed a systematic experimental and computational study to better understand how this process really works. Along the way, they found that starting with a Pd(I) bridging dimer precatalyst offers faster reaction rates and better yields than starting with a Pd(II) salt to generate the Pd(0) catalyst, as is commonly done in Buchwald-Hartwig aminations, Suzuki-Miyaura cross-couplings, and other palladium-catalyzed reactions. Colacot presented these new findings during a Division of Organic Chemistry symposium at the ACS national meeting in San Francisco.
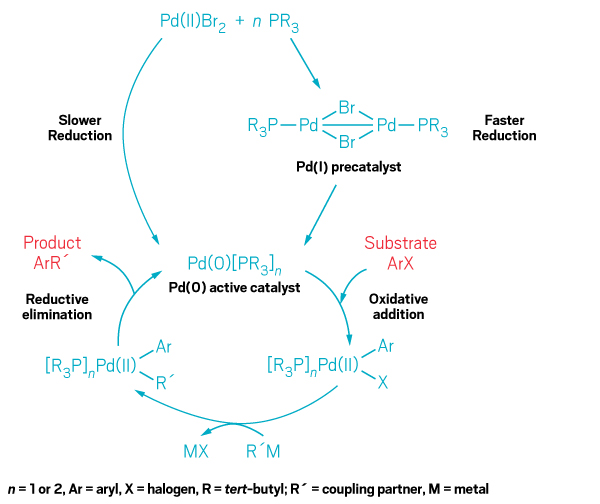
When chemists unraveled the complete mechanism of Pd(0) active catalyst formation, they found that the bromide-bridging Pd(I) phosphine precatalyst shown produces better results in cross-coupling reactions than starting with a Pd(II) precursor.
Although other researchers have previously studied the addition of phosphine ligands to a Pd(II) bromide salt and determined how a bromide-bridging Pd(I) dimer forms, a full accounting of all the molecular participants had been lacking, Colacot said. Schoenebeck, Colacot, and their colleagues determined how the ratio of Pd(II) bromide precursor to phosphine ligand, as well as the order in which the ligand and other reagents are added, dictates whether or not the Pd(I) precatalyst forms ahead of the Pd(0) catalyst. Along the way, the researchers found a key hidden intermediate species, which was visible only when the reactions were run on a gram scale. This new species, a Pd(II)Br3 dimer, turns out to be the linchpin that holds the pathway together for formation of the Pd(I) precatalyst.
Ultimately, the researchers found that starting with the preformed Pd(I) precatalyst provides better results than starting with the Pd(II) bromide salt and adding the phosphine ligand (J. Am. Chem. Soc. 2017, DOI: 10.1021/jacs.7b01110). “This implies that absolute process precision is required in catalysis to get the optimal results,” Colacot said.
The new results “are quite surprising to me, especially since our efforts studying a Pd(I) complex showed them to be very poor catalysts,” says Stanford University chemistry professor Barry M. Trost. “This paper demonstrates that the amazing subtleties of the exact structures of the active catalyst can open new windows in palladium-catalyzed processes.”
The new findings already have Bruce H. Lipshutz of the University of California, Santa Barbara, and his group thinking about ways to improve their palladium-catalyzed reaction chemistry. The symposium where Colacot made his presentation was in Lipshutz’s honor as the 2017 recipient of the HerbertC. Brown Award for Creative Research in Synthetic Methods.
The Schoenebeck and Colacot report “is a must-read,” Lipshutz said. “It’s an outstanding example of collaboration between academic and industrial groups that creates tremendous value for the catalysis community.”
Colacot said the Pd(I) precatalyst is commercially available and is being sold in kilogram amounts. In addition, other Pd(I) precatalysts based on the new information have been tested by the Schoenebeck group and are becoming available.
Organosilicon-catalyzed reaction functionalizes even notoriously inert methane, without help from precious transition metals
Unactivated alkanes are difficult to functionalize, and most catalysts that derivatize them by opening hydrocarbon C–H bonds are based on precious transition metals.
Researchers have now developed a class of intermolecular C–H arylation reactions that use catalysts made from more-abundant materials: silicon and boron. The reaction adds aryl groups to C–H bonds of simple hydrocarbons, including to the notoriously inert bonds in methane, at mild temperatures (Science 2017, DOI: 10.1126/science.aam7975).
“Alkanes are bulk components of gasoline and as such are supercheap commodities, which, if converted to functionalized compounds, would become much more valuable,” comments Jay Siegel of Tianjin University, who developed a related intramolecular reaction but was not involved in the new study. “This is an area rich in prospects, with a bright future for chemical synthetic methods development.”
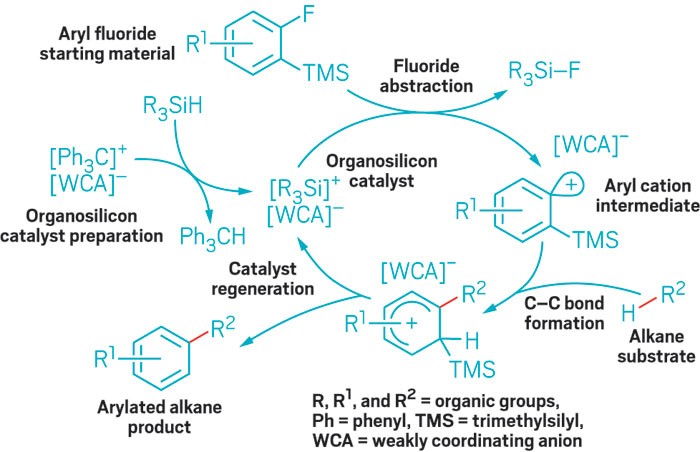
A hydrocarbon arylation reaction begins with catalyst preparation (left).
Hosea M. Nelson and coworkers at the University of California, Los Angeles, prepare the new organosilicon catalyst from an organosilane and a weakly coordinating carborane anion. The catalyst defluorinates an aryl fluoride starting material, likely generating an aryl cation intermediate that inserts electrophilically into a C–H bond of an alkane substrate to yield an arylated alkane. A key trimethylsilyl group on the aryl fluoride aids fluoride abstraction, helps the cation react quickly, and eases catalyst regeneration.
“Electrophilic reactions with methane are exceptionally rare, and the C–H functionalization of methane reveals the extraordinary reactivity of this system,” says Douglas Klumpp of Northern Illinois University, an expert on highly reactive electrophilic intermediates. The clever use of a trimethylsilyl group, he says, enabled the researchers “to tame a lion,” the aryl cation intermediate, “and that lion is able to do some very nice tricks.” However, Klumpp notes that one limitation of the chemistry is that “the aryl fluoride starting materials are expensive or difficult to obtain.”
“The chemistry isn’t ready for prime-time applications,” Nelson says. “It’s a new strategy that will hopefully fuel further study. We need to find ways to improve the reaction’s efficiency, selectivity, and substrate scope. We have filed a provisional patent and look forward to working with the chemical industry to develop practical applications.”
Chemical & Engineering News
18th International Symposium on Relations between Homogeneous and Heterogeneous Catalysis (ISHHC XVIII 2017) Sydney, Australia |
|
8th International Conference on Green and Sustainable Chemistry (GSC-8) Melbourne, Australia |
|
International Research Conference on Sustainable Energy, Engineering, Materials and Environment (SEEME) Newcastle, UK |
|
2-я Российская конференция “Графен: молекула и 2D кристалл” Новосибирск, Россия |
|
European Advanced Materials Congress Stockholm, Sweden |
|
Молодежная конференция "Применение зондовой микроскопии в научных исследованиях" Екатеринбург, Россия |
|
13th European Congress on Catalysis (Europacat 2017) Florence, Italy |
|
11th Triennial Congress of the World Association of Theoretical and Computational Chemists (WATOC 2017) Munich, Germany |
|
Международная конференция по органической химии “Байкальские чтения-2017” и Научно-практический круглый стол по вопросам развития малотоннажной химии Иркутск, Россия |
|
4th Central and Eastern European Conference on Thermal Analysis & Calorimetry (CEEC-TAC4) Chisinau, Moldova |
|
Международная конференция “Сканирующая зондовая микроскопия - 2017” (Scanning ProbeMicroscopy 2017) Екатеринбург, Россия |
|
XIX Euroanalysis Conference Stockholm, Sweden |
|
15th International Conference on Environmental Science and Technology (CEST 2017) Rhodes, Greece |
|
8th World Congress on Oxidation Catalysis “Oxidation processes: challenges and solutions” (WCOC 2017) Krakow, Poland |
|
XII European Workshop Meeting on Innovation in Selective Oxidation (ISO’17) Krakow, Poland |
|
IV International Conference“Catalysis for Renewable Sources: Fuel, Energy, Chemicals” (CRS-4) Rimini, Italy |
|
International Symposium on Synthesis & Catalysis 2017 (ISySyCat2017) Evora, Portugal |
|
XXIX Симпозиум “Современная химическая физика” Туапсе, Россия |
|
ХХ Молодежная школа-конференция по органической химии Казань, Россия |
|
Международная конференция “Возобновляемые растительные ресурсы: химия, технология, медицина” (Renewable Resources RR 2017) Санкт-Петербург, Россия |
|
II симпозиум “Современные проблемы нанокатализа” с участием зарубежных ученых (NANOCAT-2017) Киев, Украина |
|
7th IUPAC Conference on Green Chemistry (7th ICGC) Moscow, Russia |
|
IX Научно-практическая конференция “Сверхкритические флюиды: фундаментальные основы, технологии, инновации” Сочи, Россия |
|
5th International Conference on Nanotechnology and Materials Science Dubai, UAE |
|
Международный Российско-Казахстанский Симпозиум “Углехимия и экология Кузбасса” Кемерово, Россия |
|
Applied Nanotechnology and Nanoscience International Conference (ANNIC 2017) Rome, Italy |
|
Global Conference on Catalysis and Reaction Engineering (GCR-2017) Las Vegas, USA |
|
VIII конференция РХО Москва, Россия |
|
V Всероссийская научная конференция “Теоретические и экспериментальные исследования процессов синтеза, модификации и переработки полимеров” Уфа, Республика Башкортостан, Россия |
|
International Symposium on Novel Energy Nanomaterials, Catalysts and Surfaces Tokyo, Japan |
|
VII Международная конференция “Деформация и разрушение материалов и наноматериалов” Москва, Россия |
|
Всероссийская конференция по квантовой и математической химии Уфа, Республика Башкортостан, Россия |
|
II Международная научно-практическая конференция “Графен и родственные структуры: синтез, производство и применение” Тамбов, Россия |
|
II Всероссийская молодежная конференция “Проблемы и достижения химии кислород- и азотсодержащих биологически активных соединений” Уфа, Республика Башкортостан, Россия |