V Российский конгресс по катализу «РОСКАТАЛИЗ»
21-26 апреля 2025 г.
Санкт-Петербург, Россия
https://ruscat-5.tilda.ws/
Завершил свою работу V Российский конгресс по катализу «Роскатализ», который проходил в Санкт-Петербурге с 21 по 26 апреля. Он стал самым крупным за свою историю – в работе конгресса очно приняли участие более 500 человек.
Мероприятие собрало участников из 40 городов, больше всего – из Новосибирска, Москвы, Санкт-Петербурга, Томска, Казани. Программа оказалась очень насыщенной: пять пленарных лекций, девять ключевых лекций, три круглых стола, 190 устных и 128 стендовых докладов.
«Мы продуктивно провели дни “Роскатализа”: плотно поработали, послушали много интересных докладов. Любое окончание – это новое начало, и закрывая пятый конгресс, мы начинаем подготовку к шестому. Университеты будут готовить новую смену, научные сотрудники – новые исследования, промышленники – новые задачи и актуальные направления», – подытожил директор ФИЦ «Институт катализа СО РАН», академик РАН Валерий Бухтияров.
Председатель Сибирского отделения РАН, научный руководитель ФИЦ ИК СО РАН академик РАН Валентин Пармон рассказал в своем приветственном слове, что к середине 1980-х годов доля отечественных катализаторов в промышленности была доведена до 97%. «В 1990-е она упала практически до нуля, но уже сегодня российские химики обеспечили независимость по катализаторам для производства практически любых видов моторного топлива», — отметил Валентин Пармон. Он выразил пожелание, «чтобы конгресс достиг своей цели, чтобы мы продолжили обеспечивать не только технологический суверенитет России, но и заниматься тем, что поставлено во главу угла — добиваться технологического лидерства».
Санкт-Петербургский горный университет императрицы Екатерины II предоставил комфортную площадку для проведения конгресса, а его сотрудники в том числе работали над его организацией. Научный руководитель университетского Научного центра «Проблем переработки минеральных и техногенных ресурсов» д.т.н. Игорь Пягай пожелал, чтобы «Роскатализ» стал толчком к активной научной работе.
«Мы рады, что здесь собрался весь “цвет катализа”. Очень приятно видеть молодых ученых, которые двигают вперед каталитическую науку. Мы хотели бы, чтобы этот конгресс стал отправной точкой: чтобы мы могли вернуть научное преимущество, которое имели в свое время, чтобы наши ученые продвигали науку вперед, а наши производители наладили производство на самом высоком уровне. И, конечно же, чтобы он стал трамплином для исследовательской деятельности молодых ученых», – поделился он.
Заместитель руководителя Научного центра «Проблем переработки минеральных и техногенных ресурсов» к.т.н. Вячеслав Рудко отметил высокий уровень работ:
«Были представлены высококачественные фундаментальые и научно-прикладные работы — в целом полезно бывать на таких мероприятиях, которые расширяют кругозор и фокусируют междисциплинарный взгляд. Это помогает приблизиться к пониманию, почему мы сейчас находимся в процессе своего развития именно в этой точке, откуда мы начали и куда движемся».
В рамках Роскатализа прошла Молодежная школа по катализу.
Лучшие, по мнению жюри, доклады сделали:
Также были выбраны лучшие стендовые доклады:
Сайт Института катализа СО РАН
Сайт издания СО РАН «Наука в Сибири»
К 100-летию введения концепции «активных центров в катализе»
чл.-корр. РАН, проф. В.А. Лихолобов
Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск
12 февраля 1925 года в журнал Proc. R. Soc. Lond. по представлению члена Королевского Общества Э.Ф. Армстронга (Edward Frankland Armstrong), всемирно известного в то время исследователя в области превращений углеводов под действием ферментов и твёрдых катализаторов, для опубликования была принята статья доцента кафедры физической химии Принстонского университета (США) (D.Sc., Associate Professor of Physical Chemistry, Princeton University) Х.С. Тэйлора (Hugh Stott Taylor) под названием «Теория каталитической поверхности» [Hugh Stott Taylor/ A Theory of the Catalytic Surface/ Proc. R. Soc. Lond. A, (1925), 108, 105-111; doi: 10.1098/rspa.1925.0061].
В вводной части этой статьи Х.С. Тэйлор написал: «Проблема механизма каталитического действия, происходящего на поверхности твердого катализатора, может быть изучена несколькими способами. Кинетические исследования таких реакций дают важную информацию о том, каким образом осуществляется процесс. С другой стороны, внимание может быть сосредоточено не на реакциях, которые протекают на поверхности катализатора, а на свойствах самой поверхности, и по результатам таких исследований могут быть сделаны обобщения относительно процессов, которые происходят с самой поверхностью. Именно этот последний метод решения проблемы контактного катализа был применен в последние несколько лет в ходе обширной серии исследований свойств твердых каталитических поверхностей, проведенных автором и его коллегами в Принстоне» [The third Report of the Committee on Contact Catalysis of the National Research Council (by Taylor H.S., 1924); Journal of Physical Chemistry, vol. 28, p. 897, 1924; nap.nationalacademies.org/read/9561/chapter/1]. Например, было обнаружено, что образцы никелевого катализатора, приготовленные идентичными методами, проявляли значительные различия в активности при гидрировании бензола, а также в чувствительности к отравляющему воздействию небольших количеств монооксида углерода. В целом, из ряда источников были собраны данные, свидетельствующие о том, что только часть поверхности катализаторов может быть активной для осуществления катализа и, более того, что эта часть может быть значительно меньше, чем часть поверхности, способная адсорбировать газы. Было отмечено, что активные катализаторы чрезвычайно чувствительны к термической обработке и к отравлению, причём обе эти обработки изменяют как адсорбционную способность, так и каталитическую активность, но в разной степени – снижение каталитической активности часто было более выраженным, чем снижение адсорбционной способности. Такая чрезвычайная чувствительность активных металлических катализаторов к спеканию и воздействию «ядов» была объяснена Х.С. Тэйлором идеей, что каталитическая активность связана с наличием в катализаторе «незащищенных» (низко координированных) атомов металла, находящихся в углах, на рёбрах или на выступах поверхности кристаллических частиц, и что такие атомы и представляют собой «активные центры каталитической поверхности». Но оставался вопрос – а какова природа активирующего воздействия таких «активных центров» на молекулы вступающих в реакцию веществ? Принципиальный ответ на этот вопрос был дан Х.С. Тэйлором (уже в ранге профессора) в статье, вышедшей через год в том же журнале и по представлению того же члена Королевского Общества, проф. Э.Ф. Армстронга, под названием «Механизм активации на каталитических поверхностях» [Hugh Stott Taylor (1926) The mechanism of activation at catalytic surfaces/ Proc. R. Soc. Lond. A, (1926), 113, 77–86; doi.org/10.1098/rspa.1926.0140].
В этой статье обсуждались результаты экспериментальных исследований, проведённых, главным образом, в Принстоне и показавших, что количество газа (водород, азот), поглощаемого твердыми телами, иногда увеличивается с повышением температуры. Такого поведения не должно было бы происходить, если бы поглощение газа было равновесным экзотермическим процессом. Кроме того, были случаи, когда скорость поглощения увеличивалась с повышением температуры, и можно было определить соответствующую энергию активации. Для объяснения этого феномена, Х.С. Тэйлор выдвинул идею о наличии для двухатомных молекул двух типов адсорбции – слабую, но быструю молекулярную адсорбцию и более сильную, но более медленную диссоциативную адсорбцию, приводящую к образованию атомов на поверхности в результате процесса, включающего энергию активации, по величине сравнимую с энергией химических реакций. Он подчеркнул также, что требуемая степень активации зависит от конкретной системы газ/твердое вещество, и что концепция активированной адсорбции имеет фундаментальное значение для понимания каталитических процессов. В последующие годы эта концепция о наличии активированной адсорбции, хотя и вызвала интерес, была подвергнута критике как в публичных обсуждениях, так и в частных беседах [The Fourth Report of the Committee on Contact Catalysis of the National Research Council (by Hilditch, T.P., 1926); Journal of the Socie-ty of Chemical Industry, 45(27), 447-452. doi:10.1002/jctb.5000452703]. Однако в 1932 году точка зрения Тэйлора о реальности активированной адсорбции была подтверждена теоретической работой, представленной Дж. Е. Леннардом-Джонсом [J. E. Lennard-Jones. Processes of adsorption and diffusion on solid surfaces// Transactions of the Faraday Society. 1932, Vol. 28, p. 333-359; doi:10.1039/ TF9322800333], в которой было показано, что переход от физической адсорбции к хемосорбции может включать в себя энергию активации.
В течение следующих десяти-пятнадцати лет в Принстоне и других исследовательских центрах было получено все больше доказательств в поддержку концепции активированной адсорбции, а как только стали доступны реагенты, содержащие дейтерий, результаты реакций каталитического обмена между дейтерием или оксидом дейтерия и молекулами, содержащими атомы водорода, стали убедительным доказательством реальности активированной адсорбции. Обобщение этих работ представлено Х.С. Тэйлором в издании [Twelfth Report of the Committee on Catalysis, NRC, (1940): Chapter III “Activated Adsorption” https://nap.nationalacademies.org/read/21586/chapter/3#28; Chapter IV “Active Centers”; https://nap.nationalacademies.org/read/21586/ chapter/4#40], а также в авторском обзоре, получившем почётное право открыть серию изданий Advances in Catalysis [Hugh S. Taylor. The heterogeneity of catalyst surfaces for chemisorptions/ Advances in Catalysis, Volume I (1948), p. 1-26; doi.org/10.1016/ S0360-0564(08)60670-7]. В целом, история развития концепции активных центров и активированной адсорбции подробно рассмотрены Ч. Кэмбалом в мемориальной статье [Kemball Charles (1975) /Hugh Stott Taylor, 6 February 1890 - 17 April 1974/ Biogr. Mems Fell. R. Soc., 21517–547; doi.org/10.1098/rsbm.1975.0017], в которой также освещены и этапы становления Х.С. Тэйлора как учёного с мировым именем. На основе материалов, представленных в отмеченной выше публикации, а также в публикации [www.physchem.chimfak.sfedu.ru/Source/History/Persones/Taylor.html], подготовлен и нижеследующий очерк о Х.С. Тэйлоре.
ТЭЙЛОР (Taylor), Хью Стотт (Hugh Stott)
(6 февраля 1890 г. – 17 апреля 1974 г.)
 Хью Стотт Тэйлор родился в Сент-Хеленсе, Ланкашир, (St Helens, Lancashire), Великобритания. Окончил Ливерпульский университет (1910 г.) и после года работы в этом же университете в лаборатории проф. Генри Бассетта (Professor Henry Bassett, Jr) прошёл годичные стажировки в Нобелевском институте в Стокгольме в лаборатории Сванте Аррениуса, а затем – в Техническом университете Ганновера у Макса Боденштейна. Получив степень доктора философии в Ливерпульском университете (1914 г.), переехал в США, где в период 1914-1958 гг. работал в Принстонском университете (New Jersey) (профессор с 1922 г., заведующий кафедрой химии в 1926-1951 гг.). Во время Второй мировой войны и несколькими годами позже руководил рядом научно-исследовательских проектов, связанных с созданием атомной бомбы. Тэйлор сохранял интерес к исследованиям еще долгое время после того, как оставил свою профессорскую должность, уйдя на пенсию в 1958 году, и продолжал публиковаться по ряду тем, включая некоторые исследования нейлона-66 [R. Vachon, L. Rebenfeld, H. Taylor. Oxidative deg-radation of nylon 66 filaments/ Textile Res. J. 1968, 38, 716-728; DOI:10.1177/004051756803800707]. Некоторые из его последних публикаций были связаны с общими аспектами образования в колледжах и университетах.
Хью Стотт Тэйлор родился в Сент-Хеленсе, Ланкашир, (St Helens, Lancashire), Великобритания. Окончил Ливерпульский университет (1910 г.) и после года работы в этом же университете в лаборатории проф. Генри Бассетта (Professor Henry Bassett, Jr) прошёл годичные стажировки в Нобелевском институте в Стокгольме в лаборатории Сванте Аррениуса, а затем – в Техническом университете Ганновера у Макса Боденштейна. Получив степень доктора философии в Ливерпульском университете (1914 г.), переехал в США, где в период 1914-1958 гг. работал в Принстонском университете (New Jersey) (профессор с 1922 г., заведующий кафедрой химии в 1926-1951 гг.). Во время Второй мировой войны и несколькими годами позже руководил рядом научно-исследовательских проектов, связанных с созданием атомной бомбы. Тэйлор сохранял интерес к исследованиям еще долгое время после того, как оставил свою профессорскую должность, уйдя на пенсию в 1958 году, и продолжал публиковаться по ряду тем, включая некоторые исследования нейлона-66 [R. Vachon, L. Rebenfeld, H. Taylor. Oxidative deg-radation of nylon 66 filaments/ Textile Res. J. 1968, 38, 716-728; DOI:10.1177/004051756803800707]. Некоторые из его последних публикаций были связаны с общими аспектами образования в колледжах и университетах.
Большинство работ, в которые Тэйлор внес выдающийся вклад, связаны с адсорбцией и катализом, но есть и некоторые другие, которые заслуживают упоминания из-за их важности для развития других областей химии и которые демонстрируют его научный кругозор как физико-химика.
1. В Ливерпульском университете Тэйлор совместно с Г. Басеттом занимался двумя направлениями исследований: изучением соединений, образующихся при взаимодействии фосфорилхлорида с различными оксидами металлов, и фазовых равновесий в системах, содержащих нитрат кальция, воду и азотную кислоту. Интерес к фазовым равновесиям Тейлор проявлял и в течение первых лет работы в Принстоне, публикуя статьи о гидратах нитрата кальция и о влиянии различных концентраций солей калия на растворимость оксалата кальция и тартрата кальция в кислотах [Henry Bassett, jr., Hugh S. Taylor. The interaction of metallic oxides and phosphoryl chloride, alone and in the presence of certain organic compounds/ J. Chem. Soc., Trans. 1911, 99, 1402-1414; doi.org/10.1039/CT9119901402; Henry Bassett, jr., Hugh S. Taylor. LXVI. Calcium nitrate. Part I. The two-component system: calcium nitrate–water. Part II. The three-component system: calcium nitrate–nitric acid–water at 25°/ J. Chem. Soc., Trans., 1912 ,101, 576-585; doi.org/10.1039/CT9120100576].
2. В Стокгольме у С. Аррениуса он изучал влияние нейтральных солей на катализируемый кислотой гидролиз сложных эфиров и обнаружил повышение активности при добавлении хлорида калия к соляной кислоте в водном растворе. Тэйлор интерпретировал эти результаты, предположив, что при добавлении соли образуются некоторые недиссоциированные молекулы кислоты, которые обладают более высокой каталитической активностью, чем ионы водорода [W.M. Henderson, H.S. Taylor. Neutral Salt Action in Acid Solubilities/ J. Phys. Chem. 20, 1916, 664; DOI:10.1021/J150170A003]. С. Аррениус был не совсем доволен этим выводам, поскольку они несколько противоречили ожидаемым из его ионной теории. Теперь ясно, что усиление активности водных растворов кислот нейтральными солями, наблюдаемое Тэйлором, частично было обусловлено солевыми эффектами, но включало и подробно изученное позже в работах Й. Брёнстеда и К. Педерсена явление общего кислотного катализа недиссоциированными молекулами кислоты. Так, в 1914 году Тэйлор впервые предложил соотношение между каталитической константой kA кислоты в ее недиссоциированной форме и константой диссоциации кислоты, KA. На основе своих собственных и других результатов он предложил соотношение: kA/kH = KA1/2, где kH – каталитическая активность иона водорода, которое на 10 лет опередило хорошо известное соотношение kA = GAKAα для общего кислотного катализа, предложенное Й. Брёнстедом и К. Педерсеном, где GA и α являются константами для данной каталитической реакции. Работы в области кислотного катализа Тэйлор продолжал и после перезда в Принстон, но в ограниченной степени.
3. Работа Тэйлора в лаборатории М. Боденштейна показала, что воздействие α-частиц на систему водород-хлор приводит к образованию большого количества молекул хлористого водорода, M, причем на каждую образовавшуюся ионную пару, N, M/N > 1000 [Hugh S.Taylor /The interaction of hydrogen and chlorine under the influence of alpha particles./J. Am. Chem. Soc. (1915), 37, 1, 24–38; doi.org/10.1021/ja02270a003]. На этом этапе М. Боденштейн пытался интерпретировать полученные результаты в терминах последовательности ионных цепей (идея атомной цепочки, основанной на повторяющихся реакциях H + C12 → HC1 + C1, Cl + H2 → HC1 + H при воздействии на смесь H2 + Cl2 светом была высказана ранее В. Нернстом), вызванных рекциями в кластерах молекул H2 и Cl2 вокруг ионов, образующихся в результате воздействия на молекулы α-частиц. Однако окаалось, что в результате быстрой гибели ионов с переходом их в радикалы, в целом дальнейшее развитие процесса протекало по радикально-цепному механизму В. Нернста.
4. Как уже отмечалось, основные научные интересы Тэйлора во время его работы в Принстоне были связаны с исследованиями в области адсорбции и гетерогенного катализа, важнейшим результатом которых явилось выдвижение концепций «активных центров» и «активированной адсорбции» [Hugh S. Taylor. The activation energy of adsorption processes/ J. Am. Chem. Soc. 1930, 52, 12, 5298–5299; doi.org/10.1021/ja01375a509; Hugh S. Taylor. The activation energy of adsorption processes/ J. Am. Chem. Soc. 1931, 53, 2, 578-597; doi.org/10.1021/ja01353a022; Hugh S. Taylor Chemical reactions at surfaces/ Chem. Rev. 1931, 9, 1, 1-46; doi.org/10.1021/cr60032a001]. Суть этих концепций была рассмотрена выше. Однако нужно отметить, что идеи об исключительной важности для осуществления каталитического действия наличия на поверхности атомов с «нескомпенсированными» валентностями (низкокоординированных атомов) продолжают вызывать большой интерес. Во-первых, они представляют собой отход от концепции Ленгмюра, согласно которой на одно адсорбционное место приходится один вид реагента, и, как следствие, для реакции между двумя видами реагентов требуется адсорбция на дополнительном месте в соседнем участке. Поэтому при наличии низкокоординированных атомов есть основания предположить механизм гидрирования, в котором взаимодействие обоих реагентов будет происходить при адорбции их на одном и том же атоме. Во-вторых, эти идеи представляют собой пример обсуждения адсорбционного состояния частиц на металлах с точки зрения образования конкретных соединений металлов с реагентами каталитического процесса. В обоих этих аспектах – концепция двух или более соединений, связанных с одним атомом металла, и акцент на аналогии между адсорбцией и образованием химических соединений – эти взгляды Тэйлора действительно приобретают все большее значение в контексте металлоорганической химии и с обнаружением тесных параллелей, которые можно найти между гетерогенными химическими соединениями и промежуточными соединениями в гомогенном катализе комплексами металлов. Эти и другие связанные с этим аспектом вопросы рассмотрены в недавнем обзоре [Vogt, C., Weckhuysen, B.M. The concept of active site in heterogeneous catalysis. Nat Rev Chem 6, 89–111 (2022); https://doi.org/10.1038/s41570-021-00340-y].
5. Открытие дейтерия в 1932 году Х.К. Юри (H.C. Urey) и полученные вскоре после этого доказательства того, что увеличение концентрации тяжелого изотопа может быть достигнуто путем электролиза водных растворов, позволили развить новые важные направления исследований в химии. Ценность этих открытий для выяснения механизмов катализа и кинетики была немедленно оценена Тэйлором, который незамедлительно переключил свою лабораторию на “производство” тяжелой воды с таким успехом, что в течение нескольких месяцев в Принстоне было гораздо больше концентрированной тяжелой воды, чем во всем остальном мире. [Hugh S. Taylor. Heavy hydrogen—a new research tool. J. Franklin Inst. 1934, 218, 1, 1-27; doi.org/10.1016/S0016-0032(34)91376-4]. Первые опыты с D2O привели к демонстрации удивительной реакции образования дейтерированного бензола C6D6 при взаимодействии бензола с тяжёлой водой на катализаторе никель-кизельгур при температуре 473 К. [P.I. Bowman, W.S. Benedict, H.S. Taylor. The preparation and properties of benzene-d6/ J. Am. Chem. Soc. 1935, 57, 5, 960; doi.org/10.1021/ ja01308a509]. Возможность получения из тяжёлой воды дейтероводорода стимулировало направления исследований по детальному изучению механизмов активации различных водородсодержащих молекул при их адсорбции. Так, Тэйлор и его коллеги смогли показать, например, что обмен метана с дейтерием происходил при 403 К на восстановленных поверхностях никеля, в то время как активированная адсорбция была обнаружена (с помощью измерений поглощения) только при гораздо более высоких температурах. В других работах Тэйлора с сотрудниками было установлено, что обмен насыщенных углеводородов на дейтерий является гораздо более медленной реакцией, чем соответствующий обмен ненасыщенных углеводородов. Эти результаты дали также информацию о скоростях обратимой диссоциативной адсорбции алканов на поверхности металла, где было установлено, что этот процесс с участием метана протекает медленнее, чем с участием этана или пропана. Подробные исследования превращений этана на никелевом катализаторе показали, что реакция H/D-обмена легко протекает в диапазоне от 373 до 403 К, тогда как для гидрогенолиза молекулы с образованием метана требуется несколько более высокая температура от 433 до 473 К. Эти работы дали четкое представление о способности катализато-ров активировать связи С-Н с большей «готовностью», чем связи С-С, и заложили основу для многих будущих работ в этой области других исследователей.
Опыт Тэйлора в лабораторном производстве тяжелой воды в Принстоне в 1930-х годах привел его к участию в Манхэттенском проекте (Manhattan Project) в части создания тоннажного производства тяжёлой воды, необходимой в качестве замедлителя в уран-тяжеловодных реакторах получения плутония [ahf.nuclearmuseum.org/ahf/history/heavy-water-reactors/]. Тэйлор и его коллеги из Принстона помогли разработать промышленный катализатор на основе платины и древесного угля для реакции обмена между водяным паром и водородом, которая была частью процесса обогащения воды дейтерием. Результаты лабораторных экспериментов в Принстоне с небольшими количествами образцов катализатора легли в основу разработки полномасштабной установки, в которой использовались тонны катализатора и производилось более тонны тяжелой воды каждые два месяца [ahf.nuclearmuseum.org/ahf/profile/hugh-taylor/].
Научные достижения Х.С. Тэйлора были высоко оценены: в 1932 году он был избран членом Лондонского королевского общества (The Royal Society of London for the Improvement of Natural Knowledge), в период 1952-1953 гг. занимал должность президента Фарадеевского общества (Faraday Society), имя Х.С. Тэйлора носит кафедра химии в Принстонском университете.
Международная конференция ДИНАМИЧЕСКИЕ ПРОЦЕССЫ В КАТАЛИТИЧЕСКИХ СТРУКТУРАХ
27 октября - 1 ноября 2024 г.
Тюмень, Россия
https://dpcs24.org/

С 27 октября по 1 ноября 2024 года в Тюмени прошла Международная конференция «Динамические процессы в каталитических структурах». Организатором форума выступил Тюменский государственный университет (ТюмГУ) при поддержке Института катализа им. Г.К. Борескова СО РАН (Новосибирск). В число со-организаторов вошли Центр НТИ «Водород как основа низкоуглеродной экономики», ООО «Утокс», АНО Центр «Прямая связь» (Новосибирск). Мероприятие прошло в стенах Школы перспективных исследований ТюмГУ.
Конференция «Динамические процессы в каталитических структурах» (ДПКС 2024) была заявлена впервые, но у ее истока стоят два серьезных события, послужившие мощным фундаментом для появления данного форума. Это конференции «Нестационарные процессы в катализе» и «Структурированные катализаторы и реакторы». Оба эти мероприятия были в свое время посвящены наиболее перспективным направлениям интенсификации каталитических процессов: осуществлению каталитических реакций в искусственно создаваемых нестационарных условиях, а также структурированию катализаторов на различных масштабных уровнях. Конференции «Нестационарные процессы в катализе» были созданы еще в Советском Союзе в 1980-х годах известным ученым, сотрудником ИК СО РАН Ю.Ш. Матросом, с 1990 года они стали международными и проводились не только в России, но также и в США, Канаде и Японии под брендом USPC (Unsteady-State Processes in Catalysis). Мероприятие по структурированным катализаторам и реакторам было впервые проведено в виде семинара «Блочные носители и катализаторы сотовой структуры» в 1995 году под руководством другого выдающегося сотрудника ИК СО РАН З.Р. Исмагилова. Это мероприятие затем переросло в известную международную конференцию ICOSCAR (Intentional Conference On Structured Catalysts And Reactors), которая проводилась, кроме России, еще в Голландии, Италии, Китае, Испании и Германии.

Программный комитет конференции возглавили автор идеи конференции, ведущий научный сотрудник Института катализа СО РАН, д.т.н. А.Н. Загоруйко и ректор Тюменского государственного университета, к.ю.н. И.С. Романчук. В состав Программного комитета вошли известные ученые в области инжиниринга каталитических процессов и представители промышленности из многих городов России, а также зарубежные эксперты. Главой организационного комитета стал Директор Школы Естественных Наук ТюмГУ, к.х.н. А.В. Елышев, координация решения всех организационных вопросов осуществлялась сотрудницей Института катализа СО РАН Т.В. Замулиной.

Генеральный партнер конференции – ПАО «Газпром Нефть» (Санкт-Петербург), партнеры – ООО «РИОС-Инжиниринг», Омск, и ООО "КИНТЕХ ЛАБ" (Москва).
Тематика конференции включает следующие научные направления:
Теоретические аспекты создания и применения структурированных катализаторов и реакторов, а также динамических режимов в каталитических системах
Структурированные катализаторы и каталитические реакторы на их основе
Нестационарные и динамические процессы в каталитических реакторах
При формировании научной программы конференции приоритет был отдан работам, сочетающим, с одной стороны, серьезную научную основу, с другой стороны, ориентацию на решение актуальных практических задач.

В итоге в конференции ДПКС-2024 очно приняли участие 90 человек из 11 городов России (36 организаций), а также участники из Китая, Сирии и Казахстана. Онлайн доклады представили 5 участников, включая двух пленарных лекторов из Великобритании и Мексики. Опубликованы тезисы 9 заочных участников из 4-х городов России и 2-х из Баку (Азербайджан). Аудитория включала представителей академической и отраслевой науки, ВУЗов, промышленных предприятий, частных инновационных компаний. В рамках научной программы было представлено 4 пленарных лекции, 6 ключевых лекций, 42 устных докладов по пяти научным направлениям, 17 стендовых докладов, включая 2 с флэш презентациями.

Научную программу форума открыл директор Института нефтехимического синтеза им. А.В. Топчиева РАН (Москва), чл.-корр. РАН, д.х.н., профессор РАН Антон Максимов. Он представил пленарную лекцию «Эволюция катализатора в каталитическом процессе: от макродинамики к формированию катализаторов в процессе реакции». Философски подойдя к определению катализатора как вещества, ускоряющего химическую реакцию, Антон Львович по сути раскрыл динамическую природу формирования активных центров, что позволяет говорить о еще одном направлении катализа – формировании гетерогенных наноразмерных дисперсных систем in situ непосредственно в реакционной среде. Автор предложил этот подход на последующее обсуждение.
Профессор Хорхе Анчейта из Национального политехнического института Мексики, один из ведущих мировых специалистов в области инжиниринга технологий нефтепереработки, в своем выступлении затронул тему динамического моделирования современных процессов гидроочистки тяжелой нефти.
Профессор Вей Ванг, представивший Пекинский Институт химической технологии, в своей лекции осветил вопросы многомасштабного моделирования CFD реакторов с псевдоожиженным слоем катализатора.
В онлайн-режиме с докладом о плазмохимических процессах для прямой фиксации азота из атмосферного воздуха выступил профессор Евгений Ребров из Университета Уорика (Великобритания). В случае успешной разработки этот подход может совершить настоящую революцию в сфере производства азотных удобрений.

Не меньший интерес аудитории вызвали ключевые лекции:
д.х.н. Михаил Синев, ФИЦ химической физики им. Н.Н. Семенова РАН, «"Решёточный" кислород: термохимия, подвижность, реакционная способность и роль в стационарных и динамических процессах окисления»

к.х.н. Сергей Хайрулин, Институт катализа им. Г.К. Борескова СО РАН, «Блочные катализаторы сотовой структуры для решения актуальных задач защиты окружающей среды»
к.т.н. Надежда Верниковская, Институт катализа им. Г.К. Борескова СО РАН, «Моделирование динамических процессов в гетерогенных каталитических системах»
д.т.н. Руфат Абиев, Санкт-Петербургский технологический институт (Технический университет), «Микрореакторные технологии – перспективное средство для контролируемого синтеза органических продуктов и получения наноразмерных и субмикронных частиц неорганических материалов»
к.х.н. Владимир Рогожников, Потемкин Д.И., Снытников П.В., Институт катализа им. Г.К. Борескова СО РАН, «Структурированные катализаторы для приложений водородной энергетики»
к.т.н. Наталия Чумакова, Чумаков Г.А., Институт катализа им. Г.К. Борескова СО РАН и Институт математики им. С.Л. Соболева СО РАН, «Язык форм для моделирования нелинейной динамики гетерогенных каталитических реакций»
Исследователи, занимающиеся катализом, уделяют большое внимание нестационарным каталитическим процессам. В качестве причин, вызывающих интерес к этим явлениям, можно назвать, во-первых, то, что многие каталитические процессы нестационарны по своей природе – под воздействием реакционной среды во времени происходит изменение каталитической активности. Во-вторых, оказалось, что в ряде случаев искусственно созданная нестационарность позволяет конструктивно более просто решить вопросы создания оптимальных технологических режимов.

Интерес к структурированным катализаторам неуклонно растет в связи с тем, что уже давно доказаны потенциальные преимущества этого типа катализаторов. На конференции большое внимание было уделено вопросам моделирования и оптимизации структуры реакторов, а также особенностям их эксплуатации. Мероприятие собрало широкий круг экспертов, которые познакомили аудиторию с последними научными данными о разработках структурированных катализаторов и реакторов и применении их в промышленности, представили подробную информацию обо всех аспектах синтеза и применения структурированных катализаторов, таких как материалы, массоперенос, селективность, активность и стабильность, методы приготовления, предвидение каталитических свойств и характеристик. Этот всеобъемлющий обзор актуальных последних достижений обозначил пути развития как жизнеспособного решения интенсификации процессов.

В рамках общей тематики по результатам рецензирования и отбора представленных тезисов были выделены секции, привлекшие особое внимание слушателей: «Импортозамещение в сфере профессионального программного обеспечения для инжиниринга каталитических процессов» и «Разработка и инжиниринг каталитических технологий». Внедрение цифровых инструментов в практику инженерной деятельности и активное их использование давно являются одной из основных тенденций развития промышленных предприятий. Аудиторию привлекли доклады представителей компаний ООО "Центр цифровых технологий", ООО "КИНТЕХ ЛАБ" из Москвы, доцента Национального исследовательского Томского политехнического университета, к.т.н. В.А. Чузлова. По итогам дискуссии была создана межрегиональная рабочая группа по импортозамещению программного обеспечения для инжиниринга каталитических процессов.
Тематику разработки и инжиниринга каталитических технологий удачно представили компании ООО "Экострим" (Москва), ООО «РИОС-Инжиниринг» (Омск), ООО "АРСКА ТЕК" (Санкт-Петербург), а также научные сотрудники Института нефтехимии и катализа УФИЦ РАН (Уфа), и Института катализа СО РАН (Новосибирск).

Высокий интерес делегатов конференции ДПКС вызвал Круглый стол, заседание которого явилось одним из важнейших мероприятий прошедшего форума. Под общим девизом

На стендовой сессии были размещены постеры, отразившие всю тематическую направленность мероприятия. Участникам представилась возможность в неформальной обстановке продолжить обсуждение острых и актуальных вопросов, стимулированных заседаниями устных сессий и Круглого стола.
Важным нововведением на конференции ДПКС-2024 стала организация полноценного пресс-центра под руководством Светланы Албаут, итогом работы которого стало около 100 публикаций о конференции в СМИ. Из федеральных СМИ, опубликовавших материалы о конференции, можно отметить ТАСС, «Российскую газету», «Прайм», «Деловой квартал», «Накануне», «Политаналитику», «Комсомольскую правду», «Московский комсомолец», а также отраслевые специализированные издания «Нефтегаз.ру» и «Наука в Сибири».

На закрытии форума была отмечена теплая и дружественная атмосфера конференции, способствующая плодотворной и активной работе, высокий уровень представленных докладов, приятные неформальные мероприятия. Молодым ученым, победителям конкурсов на лучшие устные и стендовые доклады, были вручены дипломы. Было предложено продолжить в будущем проведение такой конференции.
Труды конференции будут опубликованы в Специальном выпуске журнала «Катализ в промышленности».
Более подробную информацию о ДПКС-2024, включая программу и сборник тезисов, можно найти на сайте https://dpcs24.org/
Материал подготовили: А.Н. Загоруйко, Т.В. Замулина
Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск
Фото: Бато Дамдинов
4 июня 2025 г.
Михайленко Максим Васильевич
Обменные взаимодействия в комплексах 3d-металлов с восстановленными производными гексаазатрифенилена
Защита диссертации на соискание ученой степени кандидата химических наук по специальности 1.4.4 - Физическая химия
Место защиты ФГБУН Федеральный исследовательский центр проблем химической физики и медицинской химии Российской академии наук, г. Черноголовка
4 июня 2025 г.
Насретдинова Гульназ Рашитовна
Медиаторный электрохимический синтез наночастиц металлов и их нанокомпозитов в объеме раствора
Защита диссертации на соискание ученой степени доктора химических наук по специальности 1.4.4 - Физическая химия
Место защиты ФГБУН «Федеральный исследовательский центр «Казанский научный центр Российской академии наук», г. Казань
5 июня 2025 г.
Ворожейкина Алеся Витальевна
Синтез и использование в катализе амфифильных сополимеров N-винилкапролактама и N-винилимидазола
Защита диссертации на соискание ученой степени кандидата химических наук по специальности 1.4.7. - Высокомолекулярные соединения
Место защиты ФГБУН Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук, г. Москва
10 июня 2025 г.
Онищенко
Ярослав Викторович
Защита диссертации на соискание ученой степени кандидата химических наук по специальности 1.4.12. - Нефтехимия
Место защиты: ФГАОУ ВО «Российский государственный университет нефти и газа (национальный исследовательский университет) имени И.М. Губкина», г. Москва
11 июня 2025 г.
Салин
Алексей Валерьевич
«Кинетика
и механизм образования фосфониевых
енолятов
и их применение в
органокатализе
Защита
диссертации на соискание ученой степени
доктора
химических наук по специальности
1.4.8. - Химия элементоорганических
соединений
Место защиты: ФГБУН «Федеральный исследовательский центр «Казанский научный центр Российской академии наук», г. Казань
17
июня 2025 г.
Дышловая Ярослава Александровна
Защита диссертации на соискание ученой степени кандидатахимических наук по специальности 2.6.9. - Технология
электрохимических процессов и защита от коррозии
Место защиты: ФГБОУ ВО «Тамбовский государственный технический университет», г. Тамбов
17
июня 2025 г.
Рыбочкин
Павел Владимирович
Защита
диссертации на соискание ученой степени
кандидата
химических наук по
специальности 1.4.14. - Кинетика и катализ
Место защиты: ФГБУН Институт органической химии им. Н.Д. Зелинского Российской академии наук, г. Москва
18
июня 2025 г.
Артемьева
Анна Сергеевна
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.14. - Кинетика и катализ
Место защиты: ФГБНУ Уфимский федеральный исследовательский центр Российской академии наук, г. Уфа
18
июня 2025 г.
Вяткин
Алексей Викторович
«Cp2ZrCl2-катализируемые реакции S- и Se-содержащих
алкинов с триметил- и триэтилалюминием
Защита
диссертации на соискание ученой степени
кандидата
химических наук по
специальности 1.4.3. - Органическая химия
Место защиты: ФГБНУ Уфимский федеральный исследовательский центр Российской академии наук, г. Уфа
18
июня 2025 г.
Иванова
Виктория Николаевна
Защита
диссертации на соискание ученой степени
кандидата
химических наук по
специальности 1.4.4. - Физическая химия
Место защиты: ФГБУН Институт неорганической химии им. А.В. Николаева Сибирского отделения Российской академии наук, г. Новосибирск
18
июня 2025 г.
Левина
Анастасия Алексеевна
Защита
диссертации на соискание ученой степени
кандидата
химических наук по
специальности 1.4.3. - Органическая химия
Место защиты: ФГБУН Институт органической химии им. Н.Д. Зелинского Российской академии наук, г. Москва
19
июня 2025 г.
Бровко
Роман Викторович
Физико-химические
характеристики трансформации спиртов
в углеводороды на поверхности цеолита
H-ZSM-5
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.4 - Физическая химия
Место защиты: ФГБОУ ВО «Тверской государственный университет», г. Тверь
19
июня 2025 г.
Дмитриева
Анастасия Алексеевна
Термодинамические
и кинетические закономерности
деоксигенации анизола в сверхкритических
условиях
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.4 - Физическая химия
Место защиты: ФГБОУ ВО «Тверской государственный университет», г. Тверь
19
июня 2025 г.
Привар
Юлия Олеговна
Криогели
хитозана, сшитые диглицидиловыми
эфирами: получение, свойства, применение
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.4 - Физическая химия
Место защиты: ФГБУН Институт химии Дальневосточного отделения Российской академии наук, г. Владивосток
25
июня 2025 г.
Бандурист
Павел Сергеевич
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.4 - Физическая химия
Место защиты: ФГБУН Федеральный исследовательский центр проблем химической физики и медицинской химии Российской академии наук, г. Черноголовка
25
июня 2025 г.
Голубцов
Георгий Викторович
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.14. - Кинетика и катализ
Место защиты: ФГБУН «Федеральный исследовательский центр «Институт катализа им. Г.К. Борескова Сибирского отделения Российской академии наук», г. Новосибирск
25
июня 2025 г.
Гуань
Пэн
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.14. - Кинетика и катализ
Место защиты: ФГБУН «Федеральный исследовательский центр «Институт катализа им. Г.К. Борескова Сибирского отделения Российской академии наук», г. Новосибирск
25
июня 2025 г.
Павлов
Александр Александрович
Спектроскопия
ЯМР парамагнитных комплексов
3d-переходных
металлов
Защита
диссертации на соискание ученой степени
доктора химических наук по специальности
1.4.4. - Физическая химия
Место защиты: ФГБУН Федеральный исследовательский центр химической физики им. Н.Н. Семенова Российской академии наук, г. Москва
26 июня 2025 г.
Паперж
Кирилл Олегович
Защита
диссертации на соискание ученой степени
кандидата
химических наук по
специальности 1.4.6. - Электрохимия
Место защиты: ФГБОУ ВО «Воронежский государственный университет», г. Воронеж
26
июня 2025 г.
Ульянова
Дарья Михайловна
Особенности
катионной полимеризации сопряженных диенов под действием алюминийорганических соединений
Защита диссертации на соискание ученой степени кандидата
химических наук по специальности 1.4.7. - Высокомолекулярные
соединения
Место защиты: ФГБОУ ВО «Волгоградский государственный технический университет», г. Волгоград
27
июня 2025 г.
Бочков
Максим Александрович
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.14. - Кинетика и катализ
Место защиты: ФГБОУ ВО «Казанский национальный исследовательский технологический университет», г. Казань
3
июля 2025 г.
Папежук
Марина Владимировна
Синтез,
строение и свойства модифицированных
гидроксиапатитов и композитных материалов
на их основе
Защита
диссертации на соискание ученой степени
кандидата химических наук по специальности
1.4.1. - Неорганическая химия
Место защиты: ФГБОУ ВО «Кубанский государственный университет», г. Краснодар
A simpler way to make complex piperidines
Route combines biocatalytic oxidation and radical cross-coupling
A new, two-step strategy for making substituted piperidines offers chemists a more efficient way to make these biologically relevant molecules. The method, which leverages enzymatic oxidation and radical cross-coupling, helps medicinal chemists quickly access structures that feature piperidine. It also sidesteps many reactions that require precious metal catalysts or expensive chiral ligands, thus making the route appealing to process chemists.
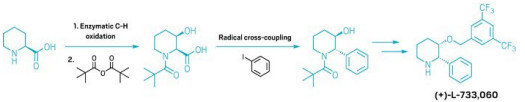
Nitrogen-containing aromatic rings like pyridine have long been popular motifs to include in drug candidates. But drugmakers looking to escape the aromatic flatlands by making molecules that reach into three dimensions have been eyeing piperidine—the unsaturated version of pyridine—as an alternative motif. The problem is that while there is well-established chemistry for adding various groups onto pyridines, there hasn’t been a convenient strategy for adding substituents to piperidines—until now.
“We thought that the combination of enzymatic functionalization and radical coupling could be leveraged to do something groundbreaking,” says Yu Kawamata of Scripps Research, who led the project along with Scripps colleague Phil S. Baran and Hans Renata at Rice University.
“Looking at various piperidines that are present in drug molecules, you see all kinds of substitution patterns,” Renata says. The diversity of arrangements didn’t point to an obvious general strategy. But the approach becomes clear if one considers using radical cross-coupling to make a carbon-carbon bond from a hydroxy group and works backward from there, he says.
The chemists started with commercially available enantiopure 2- and 3-carboxylated piperidines. Using directed evolution, they developed a series of enzymes that selectively oxidize a variety of C–H bonds on the piperidine, which provides access to a series of carboxylated piperidines with hydroxy handles. They then used radical cross-coupling reactions to forge C–C bonds on the piperidines at the sites of those hydroxy handles. Those compounds are intermediates en route to complex substituted piperidines, including the neurokinin 1 antagonist (+)-L-733,060 (shown) and the natural product swainsonine (Science 2024, DOI: 10.1126/science.adr9368).
Frank Lovering, a consultant with Thames Pharma Partners who has advocated for making more drug candidates with saturated motifs, says that using enzymes to direct functionalization of the piperidine at specific points around the ring gives chemists access to multiple molecules of interest. “However, these enzymes will need to be commercially available for this approach to be viable in the medicinal chemistry setting,” he says in an email, and currently they are not.
L.-C. Campeau, head of small-molecule process chemistry at Merck & Co., says the reaction combination demonstrates a way to eliminate inefficient steps on the way to key building blocks. “This is the type of strategy that can significantly streamline routes and enable a more green and sustainable future for chemical synthesis,” he says in an email.
A simple solution to a chlorination challenge
Iron-catalyzed reaction functionalizes less-favored positions with less fuss
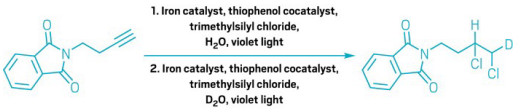
Two consecutive anti-Markovnikov hydrochlorinations of an alkyne, via the West group’s iron-catalyzed method.
Using heavy water in the second step, the researchers incorporated a deuterium atom into the product.
One of the first reactions that students learn in college-level organic chemistry is hydrochlorination—adding hydrogen chloride to a carbon-carbon double or triple bond. The classic textbook version preferentially installs the chlorine atom on the more substituted carbon in the bond, according to a selectivity principle known as the Markovnikov rule. Sticking the chlorine to the less substituted carbon is a much trickier proposition.
Traditionally, chemists have had to resort to harsh conditions, pricey reagents, or multiple synthetic steps to manage anti-Markovnikov hydrochlorination. But Julian West and his team at Rice University have devised a new way to do it with inexpensive catalysts and mild conditions (Nat. Synth. 2025, DOI: 10.1038/s44160-024-00698-z).
West says he and his group had been looking for ways to extend the iron-based radical photocatalyst system they had developed for attaching fluorinated groups to double bonds. They realized that if they used their system to generate chlorine radicals, they could do hydrochlorina-tion chemistry with flipped selectivity.
“It’s really selective for a wide range of alkenes and alkynes, but it’s also really easy to set up,” says West. Because the new approach uses inner-sphere electron transfer to generate the radicals and a thiophenol cocatalyst to shuttle hydrogen atoms, it is compatible with molecules that are easily oxidized or sensitive to ac-id—which other methods often are not. The researchers also found that doubly hydrochlorinating a triple bond with this reaction gives a product with each chlorine bound to a different carbon atom, whereas the classic method would stick them both on the same carbon.
Using heavy water, the researchers could also selectively introduce deuterium atoms along with chlorine. The weightier hydrogen isotope is useful for probing reaction mechanisms, as well as studying and tuning drugs’ metabolic stability.
To showcase the reaction’s versatility and selectivity for installing chlorine, hydrogen, and deuterium atoms in previously hard-to-access positions on molecules, the researchers reported 125 molecules made with the new chemistry, including derivatives of drugs and natural products.
Elias Picazo, an organic chemist at the University of Southern California who was not involved in the work, says it’s “definitely a synthetic advance” in terms of its scope and selectivity. He thinks that the commercially available iron catalyst will be appealing to chemists in both academia and industry.
West says he and his group aim to develop chemistry that people will find useful and approachable. The goal, he says, is to make reactions where “If somebody likes them, they could try them the same day.”
Chemical & Engineering News
| 27-28
мая 2025 г.
Лидеры России и стран СНГ: Операционная эффективность 2025 Конференция и выставка по повышению операционной эффективности в азотной, нефтехимической и нефтеперерабатывающей промышленности Красная Поляна, г. Сочи, Россия |
https://enleader.ru/events/operational-excellence-2025 ?m-message-key-id=-5071053172967997440& m-message-click-id=a32a2720-85a1-42f6- ac9f-8558198ec247& utm_source=mindbox&utm_ medium=email&utm_campaign=Opex |
|
29-30
мая 2025 г.
Лидеры России и стран СНГ: Катализаторы 2025 Конференция и выставка по катализаторам нефтепереработки и нефтехимии Красная Поляна, г. Сочи, Россия |
https://enleader.ru/events/catalysts-2025? m-message-key-id=-5071053172967997440& amp;m-message-click-id=4a757e77-2b3e-413b-bb7f- cf45438e25d4&utm_source=mindbox& utm_medium=email&utm_campaign=Opex |
|
June
2-4, 2025 5th Edition of Chemistry World Conference Rome, Italy |
https://chemistryworldconference.com/ |
|
June
2-4, 2025 21st Edition of International Conference on Catalysis, Chemical Engineering and Technology (CCT 2025) Rome, Italy |
https://catalysis-conferences.com/ |
|
22-27
июня 2025 г.
Четвертая Всероссийская конференция «Электрохимия в распределенной и атомной энергетике ЭРАЭ-2025» с. Эльбрус, Кабардино-Балкарская Республика, Россия |
https://electochem2025.tilda.ws/ |
|
23-27
июня 2025 г.
XXIX Международная Чугаевская конференция по координационной химии и VI Молодежная школа-конференция «Физико-химические методы в химии координационных соединений» г. Казань, Республика Татарстан, Россия |
http://chugaev2025.iopc.ru |
|
23-27
июня 2025 г.
Третья всероссийская школа-конференция по медицинской химии для молодых ученых (MedChemSchool-2025) г. Уфа, Республика Башкортостан, Россия |
https://xn----7sbbago2ablhuhd0e7g.xn--p1ai/medchemschool-2025/ |
|
23-29
июня 2025 г.
Школа-семинар молодых ученых «Актуальные вопросы физики функциональных материалов» молодых ученых » г. Красноярск, Россия |
https://ksc.krasn.ru/events/shkola-seminar/ |
|
June
29 – July 2, 2025 17th International Conference on Chemical and Process Engineering (ICHEAP17) Florence, Italy |
https://www.aidic.it/icheap17/ |
|
30
июня – 4 июля 2025 г.
18-я Международная конференция «Оптические методы исследования потоков» (ОМИП-2025) г. Москва, Россия |
https://omfi-conf.ru/omfi2025/OMFI-2025-Call4paper-ru.pdf |
|
June
30 – July 4, 2025 International Conference on Materials for Advanced Technologies (ICMAT) 2025 Singapore, Singapore |
https://icmat2025.mrs.org.sg/
|
|
July
13-18, 2025 32nd International Conference on Photochemistry (ICP 2025) Aachen, Germany |
https://www.icp2025.de/ |
|
July
14-18, 2025
12th World Congress of Chemical Engineering and 21st Asian Pacific Confederation of Chemical Engineering 2025 (WCCE12 & APCChE 2025) Beijing, China |
https://www.wcce12.com/
|
|
July
14-19, 2025 IUPAC World Chemistry Congress 2025 Kuala Lumpur, Malaysia |
https://iupac.org/event/iupac-world-chemistry-congress-2025/ |
|
11-15
августа 2025 г.
Пятая российская конференция «Графен: молекула и 2D кристалл» г. Новосибирск, Россия |
https://docs.google.com/forms/d/ 1umL7V6OaRT5fUsV0YnnAXozMwsBXCU1N2CC1F9NdLYc /viewform?edit_requested=true |
|
18-22
августа 2025 г.
Всероссийская научная конференция с международным участием «Современные проблемы органической химии» (СПОХ-2025) г. Новосибирск, Россия |
http://web3.nioch.nsc.ru/conf2025/index.php |
|
18-24
августа 2025 г.
VII Всероссийская школа-конференция по катализу с международным участием «Каталитический дизайн: от исследований на молекулярном уровне к практической реализации» г. Новосибирск, Россия |
https://catdesign-7.tilda.ws |
|
20-23
августа 2025 г.
Всероссийская научная конференция «Николаевские чтения 2025» г. Новосибирск, Россия |
https://conferences.niic.nsc.ru/nikolaev_2025 |
|
August
24-28, 2025 Fast Ionic Transport Systems. 86th Prague Meeting on Macromolecules Prague, Czech Republic |
https://www.imc.cas.cz/sympo/86pmm/ |
|
2-3
сентября 2025 г.
Лидеры России и стран СНГ: РАЗНОТОННАЖНАЯ ХИМИЯ 2025 Конференция и выставка по нефтехимии: проекты, технологии, оборудование, катализаторы г. Москва, Россия |
https://enleader.ru/multi-tonnagechemicals |
|
September
7-11, 2025 X Conference Modeling & Design of Molecular Materials 2025 (MDMM 2025) Wroclaw, Poland |
https://mdmm.pwr.edu.pl |
|
September
8-12, 2025 9th International Conference on Semiconductor Photochemistry (SP9) Madrid, Spain |
https://sp9.sciencesconf.org/?lang=en |
|
15-19
сентября 2025 г.
XIII Международная научная конференция «Кинетика и механизм кристаллизации. Кристаллизация и материалы нового поколения» г. Иваново, Россия |
http://crystal.isc-ras.ru |
|
15-25
сентября 2025 г.
XXXVII Симпозиум «Современная химическая физика» г. Туапсе, Россия |
http://www.chemicalphysics.ru/ |
|
September
24-26, 2025 Faraday Discussion – High-entropy alloy nanostructures: from theory to application London, United Kingdom |
https://www.rsc.org/events/detail/77926/ high-entropy-alloy-nanostructures-from-theory- to-application-faraday-discussion |
|
September
28-30, 2025 NINETEENTH Conference, Nanotechnology Based Innovation for the Environment, Energy, and Health Salerno, Italy |
https://www.aidic.it/nineteenth25/index.php |
|
29
сентября – 3 октября 2025 г.
VII семинар памяти профессора Ю.И. Ермакова «Гомогенные и закрепленные металлокомплексы в качестве катализаторов для процессов полимеризации, нефтехимии и тонкого органического синтеза» г. Казань, Республика Татарстан, Россия |
https://ermakov-2025.tilda.ws/ |
|
29
сентября – 3 октября 2025 г.
XI Международная научно-практическая конференция «Добыча, подготовка, транспорт нефти и газа» г. Томск, Россия |
http://ipc-petroleum.su/ |
|
29
сентября – 3 октября 2025 г.
Конференция по использованию рассеяния нейтронов в исследовании конденсированных сред (РНИКС-2025) г. Томск, Россия |
https://rniks2025.imp.uran.ru/ |
|
6-10
октября 2025 г. Международная конференция “Функциональные материалы” (ICFM’2025) г. Форос, Крым, Россия |
https://lomonosov-msu.ru/rus/event/9501/ |
|
6-10
октября 2025 г. VII Российская конференция (с международным участием) «Актуальные проблемы нефтехимии» г. Пятигорск, Россия |
http://conference.forenewchemistry.ras.ru/ |
|
7-9
октября 2025 г. XIV Международный российско-казахстанский симпозиум «Углехимия и экология Кузбасса» г. Кемерово, Россия |
http://www.iccms.sbras.ru/ccsymp-2025 |
|
October
12-15, 2025 3rd International Conference on Energy, Environment & Digital Transition (E2DT 2025) Palermo, Italy |
https://www.aidic.it/e2dt2025/ |
|
20-24
октября 2025 г.
II Сибирский химический симпозиум (СХС-2025) г. Томск, Россия |
https://www.irkinstchem.ru/ announcements/ii-sibirckiy-khimicheskiy- simpozium-skhs-2025 |
|
27-31
октября 2025 г.
XXVI Международная конференция по химическим реакторам (ХимРеактор-26) г. Минск, Республика Беларусь |
https://chemreactor.org/ |
|
November
3-5, 2025 4th International Online Conference on Materials (IOCM2025) Online, Switzerland |
https://sciforum.net/event/IOCM2025 |
|
24-28
ноября 2025 г.
V Всероссийская конференция с международным участием «Исследования и разработки в области химии и технологии функциональных материалов» г. Апатиты, Россия |
http://chemi-ksc.ru/m-osnovnoe/konferentsii/ |
|
December
15-20, 2025 Computational Photocatalysis – Computational Photocatalysis: Photophysics & Photochemistry at Interfaces. Machine Learning Bridges Theory and Experiment (PACIFICHEM) Honolulu, United States |
https://pacifichem2025.abstractcentral.com/submission |
|
19-20
февраля 2026 г.
Лидеры России и стран СНГ: НЕФТЕПЕРЕРАБОТКА 2026 Конференция и выставка по нефтепереработке: проекты, технологии, оборудование, катализаторы г. Москва, Россия |
https://enleader.ru/events |