17 мая 2024 года на Общем собрании Сибирского отделения РАН, посвященном 300-летнему юбилею Российской академии наук, академик Валентин Николаевич Пармон рассказал о работе Отделения в минувшем году, а также о задачах на 2024 год.
Сибирское отделение Академии наук СССР было создано по инициативе академиков М.А. Лаврентьева, С.Л. Соболева, С.А. Христиановича постановлением Совета Министров СССР от 18 мая 1957 года № 564.

В.Н. Пармон отметил, что в 2023-2024 годах деятельность членов РАН, состоящих в Отделении, и сотрудников СО РАН, а также ученых и сотрудников, работающих в организациях Сибирского региона, подведомственных Министерству науки и высшего образования России и находящихся под научно-методическим руководством СО РАН, получила признание и высокую оценку – сибирские ученые отмечены рядом престижных наград, стали лауреатами государственных и правительственных премий России, кавалерами орденов и медалей, были удостоены академических и отраслевых почетных званий.
«На сегодняшний день в СО РАН сосредоточен мощнейший потенциал – почти 11 тысяч научных сотрудников. В отделении состоят 94 академика РАН, 117 членов-корреспондентов РАН и 84 профессора РАН, – сообщил глава СО РАН. – Под научно-методическим руководством Отделения находятся 12 федеральных исследовательских центров, 68 научных и 44 организации высшего образования, подведомственные Министерству науки и высшего образования России».
В 2023 году 11 объединенных ученых советов СО РАН, в которых, по словам председателя СО РАН, сосредоточен основной научный потенциал отделения, провели экспертизу проектов и отчетов большого числа организаций.
Говоря о существенных изменениях, произошедших в жизни отечественной науки в 2023 году, председатель СО РАН отметил необходимость оперативной коррекции научно-технологических приоритетов и интенсификации исследований в тех областях, которых коснулась жесткая экономическая и технологическая блокада России со стороны Запада и Японии. «В настоящий момент безусловный приоритет для российской науки, в том числе и для Сибирского отделения РАН – восстановление технологического суверенитета по критически важным высокотехнологичным направлениям», – подчеркнул В.Н. Пармон. Он напомнил, что руководство страны в 2023 году определило десять таких направлений: это искусственный интеллект; современные и перспективные сети мобильной связи; квантовые вычисления; квантовые коммуникации; новое индустриальное программное обеспечение; новое общесистемное программное обеспечение; технологии новых материалов и веществ; водородная энергетика; системы накопления энергии; перспективные космические системы и сервисы. Кроме этого, в число важнейших задач для российского научного и научно-технического сообщества входит суверенитет страны в научном приборостроении, а также обеспечение ресурсной, экологической, биомедицинской и продовольственной безопасности страны. «Организации Сибирского отделения РАН имеют компетенции в решении всех поставленных задач, – убежден Валентин Николаевич. – Перечень этих компетенций был передан в Президиум РАН для согласования в целях использования при экспертизе госзаданий институтам, находящимся под научно-методическим руководством СО РАН. Помимо этого, роли Сибирского отделения РАН в решении проблем научно-технологического развития и обеспечении технологического суверенитета Российской Федерации было посвящено Общее собрание СО РАН, состоявшееся 18 мая 2023 года».
Основными ориентирами для научных исследований с 2024 года, по словам В.Н. Пармона, должны стать приоритеты обновленной Стратегии научно-технологического развития России: «Число основных приоритетов СНТР возросло до девяти, десятый, очень важный для нас – это фундаментальные исследования».
В качестве примера технологического лидерства России, обеспечиваемого Сибирским отделением РАН, академик Пармон привел источник синхротронного излучения поколения 4+ – ЦКП «Сибирский кольцевой источник фотонов». Строительство основных технических элементов должно завершиться к концу этого года, запуск шести пользовательских станций первой очереди – в 2025-м. Глава СО РАН подчеркнул, что большинство необходимых для СКИФ технологических решений и оборудования сибирские ученые успешно создают своими силами, в том числе аналоги импортного оборудования, поставка которого в нынешней ситуации остановлена. Важнейшим моментом в развитии этого проекта стала разработка научной программы ЦКП СКИФ, прошедшая согласование в сентябре прошлого года на заседании Президиума СО РАН, а в апреле этого — на совместном заседании Президиума РАН и Научно-технического совета Федеральной научно-технической программы развития синхротронных и нейтронных исследований и исследовательской инфраструктуры на 2019-2027 годы.
Национальный гелиогеофизический комплекс РАН В.Н. Пармон обозначил как второй пример обеспечиваемого Сибирским отделением РАН технологического лидерства России. «Этот крупнейший проект класса мегасайнс, реализуемый под эгидой Института солнечно-земной физики СО РАН (Иркутск) и госкорпорации “Ростех”, предусматривает строительство уникальных научных инструментов и установок с целью ликвидации отставания отечественной науки в области физики солнечно-земных связей и выхода на траекторию опережающего развития в фундаментальных исследованиях и решении крупных прикладных проблем, а также мониторинга ближнего космоса», – подчеркнул председатель СО РАН.
Еще один пример обеспечения Сибирским отделением РАН суверенитета в области критически важных технологий для нефтеперерабатывающего комплекса России, – запуск первой очереди крупнейшего в постсоветское время катализаторного завода, строительство которого стало возможно благодаря разработкам ФИЦ «Институт катализа им. Г. К. Борескова СО РАН». «Завод обеспечивает полную импортонезависимость России по катализаторам гидропереработки моторных топлив с возможностью выпуска дизельных топлив класса “ЕВРО-5”, арктических дизельных топлив и так далее, — акцентировал академик Пармон. — Созданные базовые российские катализаторы для нефтепереработки — катализаторы крекинга, риформинга, а также гидроочистки и гидрокрекинга вакуумного газойля — обеспечивают надежную эксплуатацию и стабильность российских нефтеперерабатывающих заводов, выпуск до 25 млн тонн в год высокооктановых компонентов бензина, авиакеросина и до 55-60 млн тонн дизельного топлива класса К5».
Также среди важнейших результатов академик Пармон упомянул разработку прямой безрастворной твердофазной технологии переработки реакторных порошков сверхвысокомолекулярного полиэтилена в пленочные нити широкой номенклатуры (ФИЦ ИК СО РАН в коллаборации) и предварительную разведку запасов марганца в Томторском месторождении (Институт геологии и минералогии им. В.С. Соболева СО РАН в коллаборации), показавшую, что оруденение марганца в корах выветривания Томторского массива имеет значительные скопления и представляет промышленный интерес.
Важной особенностью Сибирского отделения РАН его глава назвал практику инициации и реализации крупных междисциплинарных комплексных проектов. Один из них выполнен под руководством Института экономики и организации промышленного производства СО РАН и связан со стратегией комплексного освоения и развития территорий Азиатской России. Результаты его оформлены в двухтомной монографии «Новый импульс Азиатской России: источники и средства развития».
Еще один крупный интеграционный проект – «Фундаментальные основы, методы и технологии цифрового мониторинга и прогнозирования экологической обстановки Байкальской природной территории» (головная организация – Институт динамики систем и теории управления имени В. М. Матросова СО РАН, Иркутск) был завершен в прошлом году консорциумом из 16 научных организаций. Результаты проекта также обобщены в монографии и станут основой для рекомендаций по комплексному мониторингу БПТ.
«Особой гордостью Сибирского отделения является то, что мы на практике отработали систему инициирования и реализации крупных мультидисциплинарных интеграционных проектов за счет средств заинтересованных индустриальных заказчиков, а не федерального бюджета, – акцентировал В.Н. Пармон. — Ярчайший пример такого сотрудничества – “Большая норильская экспедиция” 2020-2022 годов, организованная СО РАН при поддержке ПАО “Норильский никель” в кратчайшие сроки и выполненная силами сотрудников 15 сибирских научно-исследовательских институтов». В 2022-2023 годах пять научных организаций обеспечивали полевые работы в «Большой научной экспедиции», изучавшей влияние промышленности на биоразнообразие на территории 63 тысяч км2 в Арктической зоне России. Важнейшим результатом этой экспедиции стала разработка интегрального показателя состояния экосистем в зоне воздействия объектов промышленных компаний.
Важным совместным комплексным междисциплинарным проектом СО РАН и профильных предприятий агропромышленного комплекса НСО В.Н. Пармон назвал освоение сырьевой базы сапропелей Новосибирской области (ИГМ СО РАН, ФИЦ ИК СО РАН, «Барабинский агрокомплекс», «Барабинский комбикормовый завод»). В результате многолетнего изучения сибирскими учеными сапропелей Новосибирской области и сотрудничества с индустриальными партнерами будет налажено производство широкой линейки органических удобрений. Первая очередь создаваемого предприятия предполагает выпуск 30 тыс. тонн продукции в год начиная с 3 квартала 2024 года с кратным увеличением объема выпускаемой продукции начиная с 2027-го. Кроме того, подписаны первые контракты на поставки производимых в НСО удобрений за рубеж.
В.Н. Пармон подчеркнул, что этот список интеграционных проектов СО РАН далеко не полный, более того, активно готовятся новые комплексные работы.
В числе важнейших инфраструктурных проектов председатель Сибирского отделения РАН упомянул уже находящееся в активной стадии строительство кампуса мирового уровня Новосибирского государственного университета, развитие инфраструктуры Государственной публичной научно-технической библиотеки СО РАН, проект «БиоКатТех» (ФИЦ ИК СО РАН и ФИЦ «Институт цитологии и генетики СО РАН»), развитие инновационной инфраструктуры АО «Академпарк». «Предполагается, что кампусы мирового уровня также появятся в Томске, Кемерове и других городах Сибири, – пояснил В.Н. Пармон. – Еще один важный проект – создание крупнейшего центра по производству химической продукции в Усолье-Сибирском (Иркутская область) под кураторством Иркутского института химии им. А. Е. Фаворского СО РАН».
Подытоживая свой доклад, председатель СО РАН подчеркнул, что одной из основных задач для СО РАН на 2024 год является разработка вместе с представителями федеральной и региональной власти новой редакции комплексного плана развития Сибирского отделения РАН до 2035 года с учетом приоритетов и долгосрочных планов развития СФО, а также обновление проекта развития Новосибирского научного центра «Академгородок 2.0». При этом основная цель – восстановить совместно с РАН реальные рычаги управления научными исследованиями в НИИ, в том числе возможность оперативного влияния на тематику госзаданий, утверждаемых институтам, и инициализации комплексных интеграционных исследований. Нужно решать вопрос и по восстановлению утерянной координации фундаментальных научных исследований в интересах обороны страны и международных научных связей. «Уверен, что все мероприятия плана реализации Стратегии социально-экономического развития Сибирского федерального округа до 2035 года будут выполнены. Основа уверенности в успехе намеченного — это прочность тетраэдра СО РАН, который опирается на проверенный временем треугольник Лаврентьева и единство научного сообщества Сибири», резюмировал академик Пармон.
Материалы газеты СО РАН «Наука в Сибири»
https://www.sbras.info/articles/organizaciya-nauki/predsedatel-so-ran-podvel-itogi-2023-goda
Фото Юлии Поздняковой
8-13 октября 2023 г.
Тюмень, Россия
https://chemreactor.org/

XXV Международная конференция по химическим реакторам ХимРеактор-25 была проведена в Тюмени 8-13 октября 2023 года при поддержке Правительства Тюменской области. Оператором форума выступил Западно-Сибирский межрегиональный научно-образовательный центр мирового уровня (ЗапСибНОЦ, Тюмень). В число организаторов конференции вошли Тюменский государственный университет и Институт катализа им. Г.К. Борескова СО РАН (Новосибирск). Местом проведения мероприятия стал Западно-Сибирский инновационный центр (Тюменский Технопарк).
Целью встречи ведущих экспертов в области химической и биохимической инженерии являлось представление широкого обзора последних достижений, новаторских концепций по всему диапазону прорывных инновационных исследований – от фундаментальных основ до прикладных разработок для новых приложений. По традиции программа конференции ХимРеактор была сфокусирована на фундаментальных аспектах и практическом применении каталитических процессов и химических реакторов, а также на разработке новых высокоэффективных каталитических технологий. Председателями конференции ХимРеактор-25 выступили член-корр. РАН, д.т.н., профессор Александр Степанович Носков (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск) и Генеральный Директор Западно-Сибирского межрегионального научно-образовательного центра мирового уровня Денис Владимирович Неустроев (Тюмень). Научный комитет форума возглавил академик, профессор Валентин Николаевич Пармон (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск), Программный комитет – д.т.н. Андрей Николаевич Загоруйко (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск), заместителем председателя Программного комитета выступил Директор Центра природовдохновленного инжиниринга ТюмГУ, к.х.н. Андрей Владимирович Елышев (Тюменский государственный университет, Тюмень). Впервые в рамках руководящих органов конференции был создан Научно-Индустриальный комитет (НИК), в который вошли высококвалифицированные представители промышленных компаний и бизнес-сообщества, имеющие опыт успешной работы в сфере разработки и коммерциализации новых технологий в области нефтепереработки, нефтегазохимии и химической промышленности. Миссией НИК стало использование площадки конференции ХимРеактор-25 для обсуждения развития органичного и эффективного взаимодействия науки с производством. Работу Научно-Индустриального комитета возглавил Заместитель Губернатора Тюменской области Алексей Владимирович Рейдер.
На официальном открытии конференции выступили председатель форума, заведующий отделом Института катализа им. Г.К. Борескова СО РАН, член-корр. РАН, д.т.н., профессор Александр Степанович Носков (Новосибирск), Заместитель Губернатора Тюменской области Алексей Владимирович Райдер (Тюмень), председатель Научного комитета конференции, Президент Сибирского отделения Российской академии наук, лауреат премии «Глобальная энергия», научный руководитель Института катализа им. Борескова СО РАН, академик РАН, профессор Валентин Николаевич Пармон (Новосибирск); Ректор Тюменского государственного университета, к.ю.н. Иван Сергеевич Романчук, председатель Программного комитета конференции, ведущий научный сотрудник Института катализа им. Г.К. Борескова СО РАН, д.т.н. Андрей Николаевич Загоруйко (Новосибирск).

В приветственных речах была дана статистика предстоящего мероприятия, отражена история становления конференции ХимРеактор, уходящая корнями в 60-е годы прошлого века. Начиная с небольших рабочих совещаний всероссийского уровня, постепенно мероприятие стало брендовым и широко признанным в мире как международное научное событие высокого уровня. Выступающими была представлена широкая география конференций ХимРеактор как по местам проведения форума во многих государствах мира, так и по представительству делегатов – от 38 до 50 стран практически со всех континентов. Тематика форума, обсуждаемая и формируемая членами Международного Научного комитета, постоянно расширяется и модифицируется по мере появления новых прорывных направлений. Большое внимание в программе конференций ХимРеактор уделяется технологиям природоохранного назначения, синтеза новых продуктов и высокоэффективной переработки углеводородного сырья.
Предполагалось, что в 2022 году очередная конференция ХимРеактор-25 должна была пройти в Германии. К сожалению, это оказалось невозможным по геополитическим причинам. Тем не менее, организаторы приняли решение не прерывать сложившуюся успешную традицию и предложили провести ХимРеактор-25 в 2023 году в Тюмени. Такой выбор был логичен, ведь Тюменская область известна как регион с мощной промышленностью в области нефтехимии, газохимии и переработки углеводородов, а также активно развивающейся наукой и образованием в сфере химической технологии.
В конференции ХимРеактор-25 приняли очное участие 180 ученых и специалистов из 28 городов России. С учетом онлайн участия, в том числе экспертов из шести зарубежных стран – 230 человек. По итогам отбора Программным комитетом в рамках научной программы было представлено шесть пленарных лекций, пять ключевых лекций, пять презентационных докладов компаний, 78 устных секционных докладов, 69 стендовых докладов.
Научная программа конференции ХимРеактор-25, представленная на заседаниях четырех секций, фокусировалась на следующих научных направлениях:
I. Развитие теоретических основ процессов в химических реакторах
II. Разработка химических реакторов и технологических схем реакционных процессов
III. Химические реакторы и технологии для целевых приложений
IV. Новые реакторы и технологии для приложений в топливно-энергетической сфере
В рамки Секции IV был выделен блок докладов по теме «Нефтепереработка: катализаторы и гидропроцессы».
 Одной из главных традиций конференции ХимРеактор является представление Почетной пленарной лекции, посвященной ее основателю, член-корреспонденту РАН, д.х.н., профессору Михаилу Гавриловичу Слинько. Как правило, эта лекция открывает научную программу конференции. Право на ее чтение предоставляется признанным в мире специалистам в области инжиниринга, кинетики химических реакций, основ моделирования химических реакторов. Традиционно докладчика предлагают и выбирают члены Международного Научного комитета конференции ХимРеактор. Многими членами данного комитета для представления почетной лекции памяти М.Г. Слинько была предложена кандидатура член-корр. РАН, д.т.н., профессора Александра Степановича Носкова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск). А.С. Носков, в прошлом коллега Михаила Гавриловича Слинько, является признанным в мире специалистом в области химической инженерии. Он стал автором идеи учреждения в 2008 году Почетной лекции памяти профессора М.Г. Слинько как основателя конференции ХимРеактор. Кандидатура Александра Степановича Носкова на представление Почетной пленарной лекции памяти профессора М.Г. Слинько была горячо поддержана членами Международного Научного комитета конференции ХимРеактор-25, ему выпала честь открытия научной программы форума.
Одной из главных традиций конференции ХимРеактор является представление Почетной пленарной лекции, посвященной ее основателю, член-корреспонденту РАН, д.х.н., профессору Михаилу Гавриловичу Слинько. Как правило, эта лекция открывает научную программу конференции. Право на ее чтение предоставляется признанным в мире специалистам в области инжиниринга, кинетики химических реакций, основ моделирования химических реакторов. Традиционно докладчика предлагают и выбирают члены Международного Научного комитета конференции ХимРеактор. Многими членами данного комитета для представления почетной лекции памяти М.Г. Слинько была предложена кандидатура член-корр. РАН, д.т.н., профессора Александра Степановича Носкова (Институт катализа им. Г.К. Борескова СО РАН, Новосибирск). А.С. Носков, в прошлом коллега Михаила Гавриловича Слинько, является признанным в мире специалистом в области химической инженерии. Он стал автором идеи учреждения в 2008 году Почетной лекции памяти профессора М.Г. Слинько как основателя конференции ХимРеактор. Кандидатура Александра Степановича Носкова на представление Почетной пленарной лекции памяти профессора М.Г. Слинько была горячо поддержана членами Международного Научного комитета конференции ХимРеактор-25, ему выпала честь открытия научной программы форума.
В своей пленарной лекции “Математическое моделирование гидропроцессов нефтепереработки” Александр Степанович Носков привел результаты разработки современных методов моделирования сложных многокомпонентных реакционных систем, а именно, гидропроцессов нефтепереработки: риформинга бензиновых фракций, гидроочистки дизельных фракций и вакуумного газойля, гидрокрекинга тяжелых нефтяных фракций. Общей проблемой для всех данных процессов является большое число реагентов, что осложняет разработку приемлемых для расчета реакторов кинетических моделей. В лекции А.С. Носкова показаны пути и механизмы сокращения размерности кинетических моделей без потери их прогнозной информативности для различных процессов. В докладе также были приведены результаты развития методов вычислительной гидродинамики применительно к многофазным процессам нефтепереработки. Показана возможность оптимизации конструкций каталитических реакторов для обеспечения требуемых гидродинамических режимов. В заключении лекции Александр Степанович сформулировал перспективы и задачи развития математического моделирования гидропроцессов нефтепереработки. Прежде всего, по его мнению, интерес вызывает развитие моделирования многофазных гидропроцессов на основе совместного применения методов вычислительной гидродинамики и данных кинетических исследований. Развитие современных физико-химических методов исследования каталитических процессов позволяет решить задачи изучения гидропроцессов в онлайн-режиме на основе томографии и EXAFS-спектроскопии. Александр Степанович также акцентировал, что особое внимание в ближайшие годы должно быть уделено созданию отечественных пакетов инженерных программ по моделированию гидропроцессов нефтепереработки, которые позволяют вести выбор режимов эксплуатации промышленных реакторов и получать продукты с требуемыми свойствами.
Продолжил пленарную сессию доклад директора Института экономики и организации промышленного производства СО РАН, академика РАН, д.э.н., профессора Валерия Анатольевича Крюкова совместно с соавтором к.э.н. Владимиром Витальевичем Шматом (Новосибирск). Эта лекция – “Нефтегазохимическая промышленность России – от отдельных проектов к их взаимодополняемости в рамках цепочек создания социальной ценности” была посвящена исследованиям социально-экономических проблем развития нефтегазохимического сектора в России в условиях вызовов и новых трендов.  Валерий Анатольевич констатировал, что актуальной крупной задачей в развитии нефтегазохимии стала защита национальной экономики от внешних шоков, имеющих неэкономический характер. Изменение геополитической обстановки сопровождается введением разнообразных санкций как в отношении российского экспорта (нефтепродуктов и других конкурентоспособных товаров), так и на поставки в страну разнообразного технологического оборудования и сложной химической продукции. В сложившихся условиях необходимо искать решения, связанные со значительным усилением самодостаточности отечественной экономики без ущерба для ее качества и эффективности. Одним из способов решения этой задачи профессор Крюков видит развитие средне- и малотоннажных химических производств путем достраивания существующих и создания новых технологических цепочек глубокой переработки углеводородов при привлечении программно-цифровых технологий. Таким образом создаются определенные стимулы и для роста производства, и для технологического развития. Другой значимый вызов, который сможет создать коридор будущих трендов развития нефтегазохимии, по мнению Валерия Анатольевича, связан с реализацией так называемой «зеленой повестки» в экономике, диктуемой новыми реалиями глобального характера в энергетике. Здесь развитие может получить прежде всего газохимия, в рамках которой рождаются технологии преобразования метана с получением «голубых» продуктов – водорода и аммиака, представляющих собой энергетическую альтернативу традиционному ископаемому топливу. Суммируя постулаты своей лекции, Валерий Анатольевич Крюков заключает, что ускоренное развитие нефтегазохимической промышленности возможно только при существовании четкой координации, взаимосвязанности и взаимодействия между отдельными сегментами и подотраслями, структурами власти, компаниями промышленного и инновационного бизнеса.
Валерий Анатольевич констатировал, что актуальной крупной задачей в развитии нефтегазохимии стала защита национальной экономики от внешних шоков, имеющих неэкономический характер. Изменение геополитической обстановки сопровождается введением разнообразных санкций как в отношении российского экспорта (нефтепродуктов и других конкурентоспособных товаров), так и на поставки в страну разнообразного технологического оборудования и сложной химической продукции. В сложившихся условиях необходимо искать решения, связанные со значительным усилением самодостаточности отечественной экономики без ущерба для ее качества и эффективности. Одним из способов решения этой задачи профессор Крюков видит развитие средне- и малотоннажных химических производств путем достраивания существующих и создания новых технологических цепочек глубокой переработки углеводородов при привлечении программно-цифровых технологий. Таким образом создаются определенные стимулы и для роста производства, и для технологического развития. Другой значимый вызов, который сможет создать коридор будущих трендов развития нефтегазохимии, по мнению Валерия Анатольевича, связан с реализацией так называемой «зеленой повестки» в экономике, диктуемой новыми реалиями глобального характера в энергетике. Здесь развитие может получить прежде всего газохимия, в рамках которой рождаются технологии преобразования метана с получением «голубых» продуктов – водорода и аммиака, представляющих собой энергетическую альтернативу традиционному ископаемому топливу. Суммируя постулаты своей лекции, Валерий Анатольевич Крюков заключает, что ускоренное развитие нефтегазохимической промышленности возможно только при существовании четкой координации, взаимосвязанности и взаимодействия между отдельными сегментами и подотраслями, структурами власти, компаниями промышленного и инновационного бизнеса.
В пленарной лекции директора Института теплофизики им. С.С. Кутателадзе СО РАН, академика РАН, д.ф.-м.н., профессора РАН Дмитрия Марковича Марковича (Новосибирск) “Управление процессами тепломассообмена в реакторах для химических и энергетических технологий” был представлен обзор подходов и методов для лабораторного и опытно-промышленного моделирования процессов гидрогазодинамики и тепломассообмена в аппаратах энергетических и химических технологий. Дмитрий Маркович рассказал, что для широкого класса многофазных и реагирующих потоков была исследована гидродинамическая структура и развиты методы управления спектральным составом турбулентных пульсаций и интенсивностью тепломассопереноса. Полученные результаты нашли применение при моделировании и оптимизации ряда аппаратов тепловой, гидро- и водородной энергетики. В исследованных системах обнаружен ряд новых гидродинамических явлений, таких как стационарные солитоны на поверхности жидких пленок, локальные отрывы при струйном обтекании поверхностей, вихревые образования внутри крупных нелинейных волн.  Проведен анализ современных вихреразрешающих методов математического моделирования и современных экспериментальных методик, в том числе на основе оптико-информационных технологий, для верификации и получения исчерпывающих баз экспериментальных данных, необходимых для построения оптимальных схем адаптивного управления процессами. В результате развиты научные основы, разработан ряд диагностических комплексов на основе полевых оптических методов для исследования процессов в теплофизике и энергетике. Разработанная аппаратура, в настоящее время выпускающаяся серийно, успешно эксплуатируется в ряде научных, образовательных и производственных организаций. В своей лекции профессор Маркович привел примеры разработки методов управления для ряда направлений, таких как: энергоустановки на основе сжигания углеводородного топлива (пылеугольные и газовые котлы, газовые турбины); гидравлические турбины, разделительные колонны для криогенной дистилляции; элементы тепломассообменных и реакторных систем с использованием газожидкостных потоков, включая пленки жидкости, в том числе в условиях кипения и конденсации; микрореакторные и дисперсные системы; плазмохимические технологии; применение методов искусственного интеллекта для управления интенсивностью и устойчивостью процессов в реакторных установках.
Проведен анализ современных вихреразрешающих методов математического моделирования и современных экспериментальных методик, в том числе на основе оптико-информационных технологий, для верификации и получения исчерпывающих баз экспериментальных данных, необходимых для построения оптимальных схем адаптивного управления процессами. В результате развиты научные основы, разработан ряд диагностических комплексов на основе полевых оптических методов для исследования процессов в теплофизике и энергетике. Разработанная аппаратура, в настоящее время выпускающаяся серийно, успешно эксплуатируется в ряде научных, образовательных и производственных организаций. В своей лекции профессор Маркович привел примеры разработки методов управления для ряда направлений, таких как: энергоустановки на основе сжигания углеводородного топлива (пылеугольные и газовые котлы, газовые турбины); гидравлические турбины, разделительные колонны для криогенной дистилляции; элементы тепломассообменных и реакторных систем с использованием газожидкостных потоков, включая пленки жидкости, в том числе в условиях кипения и конденсации; микрореакторные и дисперсные системы; плазмохимические технологии; применение методов искусственного интеллекта для управления интенсивностью и устойчивостью процессов в реакторных установках.
Пленарная лекция директора Института нефтехимического синтеза им. А.В. Топчиева РАН, член-корр. РАН, д.х.н., профессора РАН Максимова Антона Львовича (Москва) “Реакторные системы химического циклирования для каталитических процессов” была посвящена актуальной проблеме использования принципа химического циклирования (chemical looping) для проведения различных процессов окислительного превращения метана в ценные химические продукты.  Этот метод основан на переносе кислорода к метану путем циклического процесса с использованием твердых оксидов в качестве переносчиков кислорода («активный контакт»), чтобы избежать прямого контакта между реагентами. «Активный контакт» претерпевает многократные превращения из одной формы в другую и тем самым выступает в качестве регенерируемого катализатора. В докладе Антона Львовича обсуждались основные преимущества и недостатки этого метода для различных процессов, возможности их реакторного оформления, достижения в области создания катализаторов для процессов химического циклирования. По мнению профессора Максимова, разработка и создание подходящих катализаторов с оптимальным составом «активного контакта» – активного компонента являются ключевым вопросом в оптимизации различных химических процессов с использованием принципа химического циклирования.
Этот метод основан на переносе кислорода к метану путем циклического процесса с использованием твердых оксидов в качестве переносчиков кислорода («активный контакт»), чтобы избежать прямого контакта между реагентами. «Активный контакт» претерпевает многократные превращения из одной формы в другую и тем самым выступает в качестве регенерируемого катализатора. В докладе Антона Львовича обсуждались основные преимущества и недостатки этого метода для различных процессов, возможности их реакторного оформления, достижения в области создания катализаторов для процессов химического циклирования. По мнению профессора Максимова, разработка и создание подходящих катализаторов с оптимальным составом «активного контакта» – активного компонента являются ключевым вопросом в оптимизации различных химических процессов с использованием принципа химического циклирования.
 Профессор Хорхе Анчейтаа, Мексиканский институт нефти
(Мехико, Мексика) представил аудитории пленарную лекцию “Методика корректной оценки кинетических параметров в сложных реакционных системах”. Профессором Анчейта была предложена методология оценки кинетических параметров с использованием алгоритма Монте-Карло и анализа чувствительности. Этот подход был применен к экспериментальным данным для суспензионного гидрокрекинга тяжелой нефти с участием ионных жидкостей. Все эксперименты проводились в реакторе периодического действия при температуре реакции 430°С, давлении H2 12,3 МПа и времени реакции 0,5-6 часов. Кинетическая модель, используемая для применения предложенного подхода, и соответствующие уравнения скорости реакции были продемонстрированы профессором Анчейта на графических объектах. Было показано, что сообщаемые значения кинетических параметров можно оптимизировать, благодаря чему средняя абсолютная ошибка существенно снижается с 21,73 до 7,93%. Моделирование с использованием алгоритма Монте-Карло с различными начальными значениями параметров помогает найти наилучшее начальное предположение для дальнейшей оптимизации параметров. Прогноз выхода продукта был улучшен за счет использования средней абсолютной ошибки в качестве целевой функции, поскольку она распределялась одинаково для всех продуктов. Анализ чувствительности и статистический анализ, проведенный с использованием оптимизированных коэффициентов скорости реакции, подтвердили, что полученные результаты представляют собой оптимальные значения, которые минимизируют ошибку между расчетными и экспериментальными данными.
Профессор Хорхе Анчейтаа, Мексиканский институт нефти
(Мехико, Мексика) представил аудитории пленарную лекцию “Методика корректной оценки кинетических параметров в сложных реакционных системах”. Профессором Анчейта была предложена методология оценки кинетических параметров с использованием алгоритма Монте-Карло и анализа чувствительности. Этот подход был применен к экспериментальным данным для суспензионного гидрокрекинга тяжелой нефти с участием ионных жидкостей. Все эксперименты проводились в реакторе периодического действия при температуре реакции 430°С, давлении H2 12,3 МПа и времени реакции 0,5-6 часов. Кинетическая модель, используемая для применения предложенного подхода, и соответствующие уравнения скорости реакции были продемонстрированы профессором Анчейта на графических объектах. Было показано, что сообщаемые значения кинетических параметров можно оптимизировать, благодаря чему средняя абсолютная ошибка существенно снижается с 21,73 до 7,93%. Моделирование с использованием алгоритма Монте-Карло с различными начальными значениями параметров помогает найти наилучшее начальное предположение для дальнейшей оптимизации параметров. Прогноз выхода продукта был улучшен за счет использования средней абсолютной ошибки в качестве целевой функции, поскольку она распределялась одинаково для всех продуктов. Анализ чувствительности и статистический анализ, проведенный с использованием оптимизированных коэффициентов скорости реакции, подтвердили, что полученные результаты представляют собой оптимальные значения, которые минимизируют ошибку между расчетными и экспериментальными данными.
Завершил пленарную сессию профессор Евгений Викторович Ребров, Университет Уорик, Ковентри (графство Уэст-Мидлендс, Великобритания), Технический университет Эйндховена (Эйндховен, Нидерланды), представив лекцию “Электрификация химических реакторов неокислительной конверсии метана”. Евгений Викторович констатировал, что реакторы с неподвижным слоем традиционно имеют проблему неравномерного распределения температуры по слою. Профессор Ребров и его коллеги представили новую концепцию плазменного реактора с псевдоожижением катализатора. Вращающаяся плазма в нижней части реактора обеспечивает быструю активацию метана. В этом случае конвективный поток делает возможным быстрый перенос активных частиц, образовавшихся в плазменной секции, в зону катализатора для селективной трансформации активных частиц в желаемый продукт (этилен). Такая конфигурация обеспечивает изначально большое соотношение площади поверхности к объему, гарантируя высокие скорости тепло- и массообмена, что полезно для достижения высоких конверсий и позволяет оптимально контролировать время пребывания и распределение температуры. Ключевым вопросом данной конструкции являлось обеспечение равномерного распределения катализатора по верхней секции, предотвращая его опускание в зону плазмы за счет специального расположения перегородок в верхней секции. В своем докладе Евгений Викторович обсудил влияние эксплуатационных параметров на конверсию метана и энергоэффективность. Суммируя результаты данной работы, профессор Ребров зафиксировал, что разработана микрокинетическая модель поверхностных реакций с плазменным активированием на нанесенном медном катализаторе. Кинетическая модель была объединена с глобальной моделью плазмы для описания производительности плазменного реактора. Исследовано влияние мощности разряда, начальной концентрации CH4 и скорости входного потока на конверсию метана. Модель точно описывает картину селективности при плазмохимическом превращении метана.
Профессор Ребров и его коллеги представили новую концепцию плазменного реактора с псевдоожижением катализатора. Вращающаяся плазма в нижней части реактора обеспечивает быструю активацию метана. В этом случае конвективный поток делает возможным быстрый перенос активных частиц, образовавшихся в плазменной секции, в зону катализатора для селективной трансформации активных частиц в желаемый продукт (этилен). Такая конфигурация обеспечивает изначально большое соотношение площади поверхности к объему, гарантируя высокие скорости тепло- и массообмена, что полезно для достижения высоких конверсий и позволяет оптимально контролировать время пребывания и распределение температуры. Ключевым вопросом данной конструкции являлось обеспечение равномерного распределения катализатора по верхней секции, предотвращая его опускание в зону плазмы за счет специального расположения перегородок в верхней секции. В своем докладе Евгений Викторович обсудил влияние эксплуатационных параметров на конверсию метана и энергоэффективность. Суммируя результаты данной работы, профессор Ребров зафиксировал, что разработана микрокинетическая модель поверхностных реакций с плазменным активированием на нанесенном медном катализаторе. Кинетическая модель была объединена с глобальной моделью плазмы для описания производительности плазменного реактора. Исследовано влияние мощности разряда, начальной концентрации CH4 и скорости входного потока на конверсию метана. Модель точно описывает картину селективности при плазмохимическом превращении метана.
В рамках научной программы конференции были представлены следующие ключевые лекции:
к.ф.-м.н. Гребенников Андрей Николаевич
Российский федеральный ядерный центр – Всероссийский научно-исследовательский институт экспериментальной физики, Саров, Нижегородская область, Россия
“Развитие отечественных суперкомпьютерных технологий в РФЯЦ-ВНИИЭФ”
к.ф.-м.н. Низовцева Ирина Геннадьевна1, Чернушкин Д.В.2, Резайкин А.В.3, Свитич В.Е.1,2, Коренская А.Е.2, Микушин П.В.1, Стародумов И.О.1,3
1Уральский федеральный университет им. первого Президента России Б.Н. Ельцина, Екатеринбург, Россия
2НПО Биосинтез, Москва, Россия
3Уральский государственный медицинский университет, Екатеринбург, Россия
“Газовая ферментация – технология, меняющая правила игры. От молекулярной инженерии до биореакторов, моделирование и оптимизация процессов и аппаратов”
к.х.н. Казаков Максим Олегович
Институт катализа им. Г.К. Борескова СО РАН, Новосибирск, Россия
“Современные процессы и катализаторы гидрокрекинга вакуумных дистиллятов”
д.х.н. Зайков Юрий Павлович1, Мочалов Ю.С.2, Смирнов В.П.3, Самсонов А.А.3, Холкина А.С.1, Ковров В.А.1
1Институт высокотемпературной электрохимии УрО РАН, Екатеринбург, Россия
2АО «Прорыв», Москва, Россия
3АО НПФ «Сосны», Димитровград, Россия
“Достижения современной высокотемпературной электрохимии в атомной и водородной энергетике. Технологии и оборудование”
к.т.н. Салахов Ильдар Ильгизович, Байда А.А.1, Горшков В.О.1, Сулейманов С.М.2
1СИБУР Центр Синтеза Полиолефинов, Тобольск, Тюменская область, Россия
2СИБУР ООО «ЗапСибНефтехим», Тобольск, Тюменская область, Россия
“Газофазные процессы получения полиэтилена: современные тренды развития, технологии и реактора”

Участники конференции ХимРеактор-25 высоко оценили уровень представленных пленарных и ключевых лекций, выразив мнение, что блестящие доклады лекторов стали украшением научной программы, а высокий профессионализм, яркие запоминающиеся выступления авторов послужили стимулом для продуктивной работы мероприятия.
Интересные презентации устных докладов, представленные участниками в четырёх секциях, вызвали большой интерес аудитории, активные обсуждения, многочисленные вопросы к авторам.

Чрезвычайно привлекательной оказалась и стендовая сессия, которая включала в себя постеры не только присутствующих на мероприятии участников, но и авторов публикаций, не изыскавших возможность приехать в Тюмень, но представивших постеры через коллег, а также приславших интересные аудио и видео-презентации своих докладов. Ролики их презентаций транслировались как на стендовой сессии, так и во время перерывов заседаний устных секций

Одним из самых ярких событий программы конференции стало проведение Круглого стола Научного и Научно-индустриального комитетов “Эффективное взаимодействие науки и практики как основа достижения технологического суверенитета в химической и топливно-энергетической сферах”. Мероприятие проходило в открытом формате для всей аудитории форума.
Модератором Круглого стола выступил д.т.н. Андрей Владимирович Клейменов, ПАО «Газпром нефть» (Санкт-Петербург), со-модератором – ведущий научный сотрудник Института катализа им. Г.К. Борескова СО РАН, д.т.н. Андрей Николаевич Загоруйко (Новосибирск).
Программа Круглого стола охватывала следующие направления:
Актуальная тематика Круглого стола, отражающая органическое сочетание фундаментальных научных дисциплин с эффективным трансфером их достижений в индустриальный инжиниринг, вызвала бурные дискуссии. Обсуждалась инновационная деятельность, направленная на использование и коммерциализацию результатов научных исследований и разработок с последующим их внедрением, реализацией на рынках. В жарких дебатах приняли участие представители как научной сферы, так и промышленных компаний.

Со стимулирующими докладами на Круглом столе выступили Генеральный директор ООО "АРСКА ТЕК", к.т.н. Воловиков Артём Юрьевич (Санкт-Петербург), директор Института нефтехимического синтеза им. А.В. Топчиева РАН, член-корр. РАН, д.х.н., профессор РАН Максимов Антон Львович (Москва), руководитель Инжинирингового центра Новосибирского института органической химии им. Н.Н. Ворожцова СО РАН Заикин Павел Анатольевич (Новосибирск), главный научный сотрудник Регионального инжинирингового центра активных фармсубстанций Технопарка Санкт-Петербурга Николаев Борис Петрович (Санкт-Петербург). Обсуждение эффективных механизмов трансфера разработок и технологий из науки в индустрию, коммерциализации научных идей, инновационного развития экономики продолжалось длительное время, явно способствуя продуктивному обмену ценной информацией и установлению дальнейших контактов в среде высокопрофессиональной целевой аудитории конференции.
Большой интерес участников вызвала Выставка оборудования, контрольно-измерительных приборов, рекламной продукции и информационных материалов, организованная в рамках конференции. В ней приняли участие шесть компаний: ООО «Сигм плюс инжиниринг» (Москва), ООО «Научно-производственная фирма «Мета-хром» (Йошкар-Ола, Республика Марий Эл), ООО «НКЦ «ЛАБТЕСТ» (Москва), ООО “ПраймКемикалсГрупп” (Мытищи, Московская область), ООО «ЭЛЕМЕНТ» (Екатеринбург), ООО «Сервис-центр «ХромоСиб» (Омск). Экспонентами компаний были представлены презентационные доклады.

Закрытие форума провел председатель Программного комитета XXV Международной конференции по химическим реакторам ХимРеактор-25, ведущий научный сотрудник Института катализа им. Г.К. Борескова СО РАН, д.т.н. Андрей Николаевич Загоруйко. Он представил статистику мероприятия, предоставил слово председателю конференции, член-корр. РАН, д.т.н., профессору Александру Степановичу Носкову и представителю Тюменского государственного университета, к.х.н. Андрею Владимировичу Елышеву. Молодым ученым, победителям конкурсов на лучшие устные и стендовые доклады, были вручены дипломы.

Труды конференции публикуются в Специальных выпусках журналов «Катализ в промышленности» и «Теоретические основы химической технологии».
По окончании конференции ее делегатам был предложен тур в сказочный город Тобольск, по дороге в который они посетили село Абалак, знаменитое великолепным видом на Иртыш, где на берегу этой живописной реки находится Свято-Знаменский мужской монастырь. Перед посещением музеев и главной архитектурной доминанты Тобольска – Кремля со сверкающими куполами Софийско-Успенского кафедрального собора и белоснежными башнями с ангелами на шпилях – участники нанесли визит на нефтехимическое производство ООО «ЗапСибНефтехим». Здесь они посетили крупнейшую промышленную площадку города, поближе познакомились с нефтехимическими и экологическими проектами компании СИБУР. При общении с технологами компании участникам конференции казалось, что они присутствуют на заседании второго раунда Круглого стола – удалось обсудить много важных и интересных вопросов и тем. В целом поездка в Тобольск оказалась незабываемым путешествием, хотя и немного грустным перед прощанием с конференцией ХимРеактор-25…

Участники XXV Международной конференции по химическим реакторам ХимРеактор-25 отметили высокий уровень ее организации, теплую и дружественную атмосферу, традиционно характерную для этого форума. Комфортная и творческая среда послужила достойной площадкой для обмена научными идеями, для обсуждений и споров, а неформальные мероприятия оставят приятные воспоминания от общения друг с другом и надежду на встречу на следующих конференциях ХимРеактор.
Следующая конференция ХимРеактор-26 запланирована на 2025 год. Традиционно место проведения конференции Научный комитет определяет на конкурсной основе из предложений, поступающих от активных институтов, университетов и компаний. Сбор предложений по месту проведения конференции ХимРеактор-26 уже начался.
Материал подготовили:
А.С. Носков, А.Н. Загоруйко, Т.В. Замулина
Институт катализа им. Г.К. Борескова СО РАН, г. Новосибирск
Фотографии с конференции: Светлана Албаут
Групповое фото: Екатерина Кармацкая
Photochemistry unleashes a one-two radical punch for efficient ring synthesis
Iridium and nickel catalysts cooperate to install nonaromatic rings in drug molecules
Alight-driven reaction sequence that bolts nonaromatic rings onto het-eroarenes, such as pyridines, offers a rapid route to medicinal molecules that would be troublesome to make using existing methods (Nature 2024, DOI: 10.1038/s41586-024-07181-x).
Adding saturated sp3 carbons into drug candidates can improve properties such as solubility and target-binding affinity. But fusing nonaromatic rings to heteroarenes can be laborious, leaving this type of structure underrepresented in medicinal chemistry compound libraries.
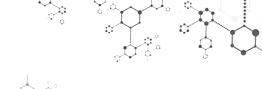
A team led by David W. C. MacMillan at Princeton University has now developed a one-pot reaction system that creates these fused ring sys-tems using simple and readily-available radical precursors such as diols and bromoalcohols. Collaborators at Janssen Research and Development are already using it in their medicinal chemistry work.
The process initially uses blue light and an iridium photocatalyst to generate a carbon radical from the precursor. A nickel catalyst then helps that radical to couple to the heteroarene. MacMillan’s group reported this strategy in 2021, but the new system now adds a second coupling to the mix.
After the precursor has been tethered to the arene, the iridium removes its other functional group to generate another carbon radical. This locks on to an adjacent point on the heteroarene to close up a nonaromatic ring.
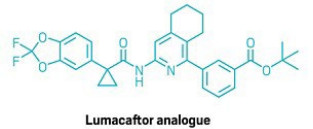
The team showcased more than 50 examples with a range of heteroarenes—including pyridines, quinolines, and pyrimidines—and created analogues of drugs such as the cystic fibrosis treatment Lumacaftor (shown). The reaction does not interfere with other functional groups in the molecules, so medicinal chemists can use it to modify complex molecules in the final stages of a synthesis. “We were surprised how well-behaved the whole thing was,” MacMillan says.
He adds that the reaction could be applied to a much wider range of coupling partners than diols or bromoalcohols. “You can make radicals from almost anything now,” MacMillan says. “So this strategy should work with almost any two functional group precursors that you want to use.”
Thermoset plastic made from wood waste catalyzes its own degradation
Scientists design tough cross-linked material that can be chemically recycled under mild conditions
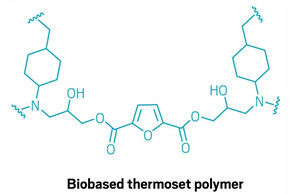
A future when tough polymer resins and composites such as those found in wind turbine blades could be made entirely from plant-based, recyclable components just got a little closer. Researchers led by Katalin Barta at the University of Graz have designed a new thermoset polymer, derived from wood, that can be broken down in methanol with no added catalyst (Science 2024, DOI: 10.1126/science.adj9989).
Thermoset polymers have cross-linked networks that make them exceptionally tough and useful materials. But they are also practically impossible to break down or recycle. And epoxyamine resins, one of the most common thermoset plastics, are typically made using bisphenol A (BPA), which is an endocrine disruptor.
Plenty of researchers have tried making degradable thermoset plastics by incorporating functional groups whose bonds can be severed by a catalyst or other external trigger. Barta and coworkers designed their new biobased epoxyamine polymer similarly, with easily cleaved ester groups in the polymer backbone. But the polymer turned out not to need an external catalyst to break it down. “The fact that it catalyzes its own degradation was definitely serendipity,” Barta says. “We didn’t hope for such a wonderful effect.” Research in Barta’s group focuses on developing useful chemical building blocks from plant-based feedstocks such as lignin, a waste product from papermaking. To make the recyclable resin, the researchers combined lignin-derived 4,4′-methylenebiscyclohexanamine (MBCA) with an epoxy component made from 2,5-furandicarboxylic acid and glycidol, both of which also come from plant-based sources.
The polymer has physical properties matching those of petrochemical-based materials, including its glass transition temperature. It’s also resistant to most solvents, including acidic and basic aqueous solutions, acetone, and dichloromethane. But Barta and Xianyuan Wu, who worked on the project as a graduate student, were surprised to discover that the material started breaking down and dissolving when soaked in methanol.
The researchers determined that hydrogen-bonding interactions between the ester, amine, and methanol activate the carbonyl and initiate a trans-esterification process that breaks up the polymer network. It takes about 48 h at 70 °C for the polymer to degrade to the point at which the methyl ester of 2,5-furandicarboxylic acid can be recovered in 90% yield. The researchers could also recover the MBCA and glycidol pieces can be separated out, so every component of the polymer is recyclable to some degree.
Eugene Y.-X. Chen, a polymer chemist at Colorado State University who was not involved in the work, calls it “a superb example of demonstrating that biobased polymers, even in a robust cross-linked network form, can also exhibit recyclability advantages.” He says it was “truly remark-able” to see an epoxy-amine resin that can be depolymerized under relatively mild conditions with no external catalyst.
The team is still working to optimize the recycling process and expand the scope of recyclable epoxy-amine polymers, but “there is definitely a potential there,” Barta says. She hopes to start looking into industrial partnerships and applications for the material soon.
Boldly going where no C–H activation has gone before
Chiral catalyst reaches remote bonds on cyclic compounds
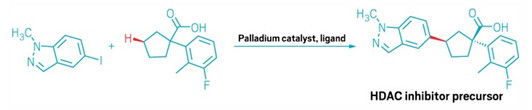
The researchers used their method to shave the synthesis of a
histone deacetylase (HDAC) inhibitor from 10 steps to 2.
C–H activation is the art of snipping specific bonds between carbon and hydrogen atoms on an organic molecule to graft a new functional group in place of the relatively unreactive H atom. Over the years, chemists have devised increasingly effective and selective ways to do C-H activation. But some bonds remain tantalizingly out of reach.
Jin-Quan Yu and his team at Scripps Research in California have been working for over 20 years to push the boundaries of which C–H bonds are possible to alter. In a new Science paper, they describe how they ste-reoselectively snipped a C–H bond and added aryl groups to the position three or four atoms away from the carbonyl carbon of cyclic carboxylic acids (2024, DOI: 10.1126/science.ado1246).
Chemists have gotten pretty good at stereoselectively adding atoms one or two spaces away from carbonyl groups, which coordinate to metal catalysts to direct them to the correct place to carry out the reaction. Proximity to a carbonyl group also makes a C–H bond easier to break. The ability to also add a stereocenter three carbons away, in the γ position, is “very enabling” for synthesis, Yu says. He and his team pub-lished a nonstereoselective γ-arylation reaction last year; introducing chirality was the natural next step (Nature 2023, DOI: 10.1038/s41586-023-06000-z).
The pursuit of reactions that can access remote C–H bonds with perfect control over the product’s 3D structure has a somewhat fraught history. In 2019, Frances Arnold’s group at the California Institute of Technology published a Science paper detailing engineered enzymes that produced chiral four-, five-, and six-membered lactam rings through C–H amidation. In 2020, a team from Hokkaido University reported, also in Science, a method using an iridium catalyst to install boron groups at the γ position of amide and ester compounds. Both papers were retracted because the results couldn’t be replicated.
The reaction Yu and his team developed relies on a palladium catalyst with a chiral oxazoline-pyridone ligand that reaches across the ring from the carboxylic acid to the γ position. It can attach a range of aryl halides to acids with five-, six-, and seven-membered rings, creating two new stereocenters in the process. The team also devised a slightly modified catalyst that can access C–H bonds four carbons away from the acid.
“I think it is really spectacular,” Huw Davies, an organic chemist at Emory University, says in an email. The Yu group has a long track record of making big advances in C–H activation methods, he adds, and this work clearly builds on those past insights. He’s interested to see what other functionality will become possible to install stereoselectively to the γ position.
Yu says he has filed a patent for the reaction and has established a start-up, Architect Therapeutics, that is based on using C–H activation catalysts to build novel scaffolds for drug discovery. Identifying how to expand the list of atoms and functional groups that can be attached to the γ position is also on his to-do list, he says. Ideally, he says, an effective C–H activation method means “you could replace the C–H bond with anything you want.”
Chemical & Engineering News
|
June
17-21, 2024 12th European Conference on Solar Chemistry and Photocatalysis: Energy and Environmental Applications (SPEA12) Belfast, UK |
https://www.ulster.ac.uk/conference/spea12 |
|
17-21
июня
2024 г.
XII Международная конференция “Механизмы каталитических реакций” (MCR-XII) г. Владимир, Россия |
https://mcr-12.tilda.ws/ |
|
June
24-28, 2024 85th Prague Meeting on Macromolecules - Polymers for Sustainable Future Prague, Czech Republic |
https://www.imc.cas.cz/sympo/85pmm/ |
|
June
30 – July 3, 2024 Industrial Biotechnology International Conference (IBIC 2024) Bologna, Italy |
https://www.aidic.it/ibic2024/ |
|
1-5
июля 2024 г.
XXIV Международная конференция по химической термодинамике в России г. Иваново, Россия |
http://rcct.isc-ras.ru/ |
|
2-4
июля 2024 г.
IV Международная конференция «Горячие точки химии твердого тела: ориентированные фундаментальные исследования» (HTSSC-2024) г. Новосибирск, Россия |
http://www.solid.nsc.ru/htssc2024/ |
|
2-6
июля 2024 г.
3-я международная конференция «Физика и химия горения и экстремальных процессов» (ComPhysChem'24) г. Самара, Россия |
http://comphyschem.ssau.ru/en/ |
|
9-12
июля 2024 г.
V Всероссийская конференция «Физико-химическая биология» г. Новосибирск, Россия |
https://www.pcb2024.ru/ |
|
July
10-12, 2024 International Symposium on Catalytic Chemistry of C1 Molecules Lille, France |
https://www.c1chem.org/ |
|
July
10-12, 2024 International Symposium SPECTROCAT 2024 – Advanced spectroscopies and operando characterizations for catalysis Caen, France |
https://www.lcs.ensicaen.fr/spectrocat-2024/?lang=en |
|
July
10-13, 2024 4th Frontiers in Photochemistry Conference Lisbon, Portugal |
https://www.fusion-conferences.com/conference/163 |
|
July
14-19, 2024 18th International Congress on Catalysis (18th ICC) Lyon, France |
https://www.icc-lyon2024.fr/ |
|
July
24-26, 2024 21st International Conference on Advanced Nanomaterials Aveiro, Portugal |
https://www.advanced-nanomaterials-conference.com/ |
|
July
24-26, 2024 6th International Conference on Green Chemistry and Sustainable Engineering (GreenChem-24) Lisbon, Portugal |
https://greenchem-europe.eu/ |
|
August
16-17, 2024 International Conference on Chemistry and Chemical Engineering (ICCCE-2024) Singapore, Singapore |
https://unitedresearchforum.com/chemistry-conference/ |
|
August
17-19, 2024 Carbon Chemistry World Conference, CCWC 2024 Barcelona, Spain |
https://www.carbonchemistryconference.com/ |
|
August
25-29, 2024 27th International Congress of Chemical and Process Engineering (CHISA 2024) Prague, Czech Republic |
https://2024.chisa.cz/ |
|
September
1-5, 2024 13th International Symposium on Heterogeneous Catalysis (13 ISHC) Burgas, Bulgaria |
https://13symp.sciconf.eu/index.php |
|
September
2-6, 2024 XIII International Conference on Chemistry for Young Scientists “Mendeleev 2024” Saint Petersburg, Russia |
https://events.spbu.ru/events/mendeleev-2024 |
|
September
2-6, 2024 25th Jubilee Annual Conference on Materials Science (YUCOMAT 2024) Herceg Novi, Montenegro |
https://www.mrs-serbia.org.rs/index.php/yucomat/yucomat-2024-xiii-wrtcs |
|
4-6
сентября 2024 г.
IX Всероссийская научно-техническая конференция молодых ученых «Перспективы создания и применения конденсированных высокоэнергетических материалов» г. Бийск, Россия |
www.ipcet.ru conf.ipcet@rambler.ru |
|
September
10-12, 2024 Beilstein Organic Chemistry Symposium – Main-group Chemistry for Modern Catalysis and Synthesis Limburg an der Lahn, Germany |
https://www.beilstein-institut.de/en/symposia/main-group-chemistry/ |
|
10-13
сентября 2024 г.
III Всероссийская конференция «Физико-технические проблемы добычи, транспорта и переработки органического сырья в условиях холодного климата» г. Якутск, Россия |
https://ipng.ysn.ru/iii-vserossijskaya-konferencziya-fiziko-tehnicheskie-problemy-dobychi-transporta-i-pererabotki-organicheskogo-syrya-v-usloviyah-holodnogo-klimata-posvyashhennaya-25-letiyu-instituta-probl/ |
|
16-20
сентября 2024 г.
XIII Всероссийская конференция (с международным участием) «Химия твёрдого тела и функциональные материалы - 2024» г. Санкт-Петербург, Россия |
http://ssc-fm2024.ru/ |
|
16-26
сентября 2024 г.
XXXVI Симпозиум «Современная химическая физика» г. Туапсе, Россия |
https://www.chemicalphysics.ru/ |
|
September
17-20, 2024 4th International Conference on Fundamentals and Applications of Cerium Dioxide in Catalysis (Ceria 2024) Portorose, Slovenia |
https://ceria2024.chem-soc.si/ |
|
23-27
сентября 2024 г.
XIII Международная конференция «Химия нефти и газа» г. Томск, Россия |
http://petroleum-chemistry.ru/ |
|
30
сентября – 3 октября 2024 г.
IX Международный симпозиум «Химия и химическое образование» г. Владивосток, Россия |
https://www.iscce.ru/ |
|
September
30 – October 3, 2024 VIII International School-Conference for Young Scientists “Catalysis: from Science to Industry” (CatConf2024) Tomsk, Russia |
http://catconf.tsu.ru |
|
3-5
октября 2024 г.
V Научно-технологический симпозиум «Гидропроцессы в катализе» (HydroCat 2024) г. Сочи, Россия |
https://sts-5.tilda.ws/ |
|
7-12
октября 2024 г.
XXII Менделеевский съезд по общей и прикладной химии Федеральная территория «Сириус», Россия |
https://mendeleevcongress.ru |
|
15-17
октября 2024 г.
XIII Международный российско-казахстанский симпозиум «Углехимия и экология Кузбасса» г. Кемерово, Россия |
http://www.iccms.sbras.ru/ccsymp-2024 |
|
October
18-22, 2024 10th IUPAC International Conference on Green Chemistry Beijing, China |
https://iupac.org/event/10th-iupac-international-conference-on-green-chemistry/ |
|
October
21-25, 2024 5th International Conference on Materials Science & Nanotechnology Athens, Greece |
https://materialsconference.yuktan.com/ |
|
24-25
октября 2024 г.
Всероссийская научно-практическая
конференция с международным участием «Водород. Технологии. Будущее» г. Пермь, Россия |
https://htf.tpu.ru/ |
|
28
октября – 1 ноября 2024 г.
Международная конференция «Динамические
процессы в каталитических структурах»
(ДПКС 2024) г. Тюмень, Россия |
https://dpcs24.org/ |
|
November
4-8, 2024 5th International Conference on Emerging Advanced Nanomaterials (ICEAN 2024) Newcastle, Australia |
https://www.newcastle.edu.au/research/centre/gican/icean-2024 |
|
11-14
ноября 2024 г.
III Международная конференция “Использование синхротронного излучения для исследования катализаторов и функциональных материалов” г. Томск, Россия |
https://srtcfm-2024.tilda.ws/ |
|
11-15
ноября 2024 г.
Всероссийская конференция им. академика В.И. Овчаренко «Органические радикалы и органическая электрохимия: фундаментальные и прикладные аспекты» г. Новосибирск, Россия |
http://or-2024.tomo.nsc.ru/index.php?lang=ru |
|
November
15-19, 2024 г.
IX International Conference of Young Scientists “Synthesis of Electrode Materials” Moscow, Russia |
https://crei.skoltech.ru/cest/conference-of-young-scientists-2024/ |
|
November
17-22, 2024 9th International Symposium on Practical Surface Analysis (PSA-24) Pusan, Korea |
https://surfaceanalysis.kr/PSA/PSA24/ |
2025 |
|
|
February
13-15, 2025 International Meet & Expo on Chemical Engineering and Catalysis Dubai, United Arab Emirates |
https://chemcatmeet.org/ |
|
21-26
апреля 2025 г.
V Российский конгресс по катализу «РОСКАТАЛИЗ» г. Санкт-Петербург, Россия |
https://ruscat-5.tilda.ws/ |