Тел.: +7 (383) 330-67-71, факс: +7 (383) 330-80-56, E-mail: bic@catalysis.ru
630090, Россия, Новосибирск, пр-т Ак. Лаврентьева, 5
Тел.: +7 (383) 330-67-71, факс: +7 (383) 330-80-56, E-mail: bic@catalysis.ru
630090, Россия, Новосибирск, пр-т Ак. Лаврентьева, 5
Head-to-tail macrocycle synthesis
Multifunctional catalyst stereoselectively ties up loose ends
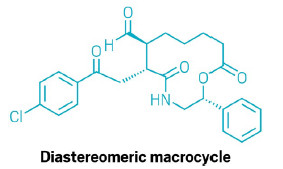
Making macrocycles is an art of many compromises. The extreme conformational flexibility of long linear molecules needed to make rings containing 12 or more atoms means that ring closure is a battle against entropy and competing dimerization. Successful ring closure is often only achieved at high dilution and stereocontrol, either embedded within the linear precursor or installed by subsequent modifications. But de-spite these difficulties, macrocyclic structures are attractive as pharma-ceuticals because they can combine the specificity of protein-based drugs with the stability of small molecules.
Fortunately, these giant rings are about to become a lot easier to access. Helma Wennemers and her team at ETH Zurich have developed a bifunctional peptide catalyst which neatly and stereoselectively produces 12-to-18-membered rings from long linear molecules under mild and scalable conditions (Science 2026, DOI: 10.1126/science.aec8992).
At one end of the peptide unit, a chiral organocatalyst forms a covalent bond with the end of the substrate. A short sequence of three amino acids—acting as a chiral spacer—then separates the first catalytic end of the molecule from the second active site: a carboxylic acid motif that activates the linear molecule via hydrogen bonding. Consequently, the head and tail of the substrate are not only primed for reaction but also held in close proximity, reducing the entropy barrier.
“The neatest bit is just how many different jobs that catalyst is doing,” says Will Unsworth, a synthetic organic chemist at the University of York who wasn’t involved in the work. “It’s activating the two ends, it’s got the chirality that’s controlling both the enantio- and diastereoselec-tivity, and it’s pre-organizing the system to favor macrocycle formation versus intermolecular reactions.”
The ETH team quickly established a set of optimized conditions for cyclizing linear substrates comprising an aldehyde ‘head’ and a Michael acceptor ‘tail.’ The researchers found they could easily generate 12-to-18-membered macrocycles, with rings of 13 or more atoms forming with >90% yield and near-perfect enantio- and diastereoselectivity. The cyclization also tolerated a variety of substrate modifications including electron donating and withdrawing groups on the Michael acceptor, and amides, esters, and strain-inducing alkynes within the tethering chain.
However, to have genuine application within pharmaceutical synthesis, predictable compatibility with chiral substrates is essential. The team therefore prepared a chiral linear precursor and tried the macrocyclization reaction with both enantiomers of a bifunctional catalyst. The catalyst dominated the stereochemistry of the crucial bond-forming steps, and the two reactions formed a complementary pair of diastereomeric macrocycles. One of the pair contained the macrocyclic core of robotnikinin, an inhibitor of the Sonic Hedgehog protein, which is implicated in some cancers.
For Unsworth, though, possibly the greatest strength of this work is the novelty and generality of Wennemers’ approach. “The idea of using a small peptide to template two reactive groups together to make a large ring, but also control stereoselectivity, is likely to lead to other synthetic applications,” he says. “You could easily imagine switching out the amine or the acid for some other functional groups and seeing whether this concept is applicable to other activating groups and bond formations.”
Hydrocarbon COFs go crystalline
A simple synthetic tweak allows building blocks to arrange themselves in a well-ordered carbon scaffold
Researchers have prepared flat, porous sheets of just carbon and hydro-gen arranged in a hexagonal network. The 2D material is the first crystalline covalent organic framework (COF) with an all-carbon backbone (J. Am. Chem. Soc. 2026, DOI: 10.1021/jacs.5c22787). The COF exhibits photoluminescent properties that are far superior to those of its amorphous counterpart.

A new covalent organic framework is made of vast crystalline sheets
of hydrocarbons with large pores throughout.
Growing crystalline COFs is more art than science. Much of the process depends on bonds between building blocks forming correctly—or break-ing and reforming if they didn’t initially join up the right way. “This reversibility is essential in all crystallization,” says Seungkyu Lee, a nanomaterials scientist at the University of Hong Kong (HKU) and the senior author of the new study.
But carbon-carbon bonds are too strong and irreversible. “If miscon-nected, there is no way you can disassociate and correct the error,” Lee explains. Hydrocarbon COFs have been prepared previously in amorphous forms—configurations that lack long-range order. To make crystalline COFs, scientists had to include noncarbon atoms in the materials’ backbones to allow for reversible bonds, such as imine bonds.
Lee’s group pulled off the hydrocarbon synthesis using the classic olefin metathesis reaction but with an additional ingredient that helps reverse C–C bonds: a free-floating olefin in the solution. The extra olefin helps the metathesis catalyst break up malformed bonds in the growing COF, giving them a second chance to form correctly.
To test their idea, the researchers mixed a triangular hydrocarbon building block, a Grubbs metathesis catalyst, and the common olefin transstilbene at 60 °C. Their method not only produced crystals of the COF—dubbed HKU-50—but even converted amorphous HKU-50 to its crystalline form.
The crystalline version of HKU-50 is four times as luminescent as the amorphous form. Lee’s collaborators, David Phillips at HKU and Nak Cheon Jeong at Daegu Gyeongbuk Institute of Science and Technology, expect the 2D material to be useful for flexible electronics or chemical sensing. While they pursue new applications for HKU-50, Lee’s group plans to use the new synthetic strategy to prepare more hydrocarbon COFs, including 3D ones.
A radical new way to alkylate aromatic rings
Chemists made a photocatalytic discovery when their control reaction led to an unexpected product
Chemists in Erwin Reisner’s group at the University of Cambridge have discovered a light-activated way to attach saturated carbon groups to the most electron-poor sites on aromatic rings without a transition metal catalyst (Nat. Synth. 2026, DOI: 10.1038/s44160-026-00994-w).
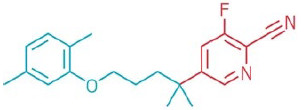
Gemfibrozil derivative
Researchers from the University of Cambridge used a new light-activated reaction
to modify several drug molecules, including the lipid-regulating medication gemfibrozil.
The researchers dubbed it the “anti-Friedel-Crafts” reaction, because its selectivity is opposite to that of the nearly 150-year-old aromatic substitution reaction, which favors electron-rich sites on electron-rich rings.
“This is far milder, and it does something completely different to established methods,” says David Vahey, the PhD student who led the experimental work. And it all came about thanks to an unexpected product from a control reaction.
Vahey had been working on aldehyde-ketone coupling with a semiconductor photocatalyst, an offshoot of the group’s broader focus on solar-powered chemistry. When he ran a control reaction without any photocatalyst, he found that the starting materials still reacted—and formed an aromatic alkylation product rather than the expected coupling product. Intrigued, the researchers decided to investigate the mechanism further.
The team determined that the reaction was mediated by a light-generated radical ion pair composed of a bulky amine base and a redox-active phthalimide ester bearing the alkyl group that would ultimately transfer to the product.
The Cambridge team joined forces with computational chemist Max García-Melchor of CIC energiGUNE and Trinity College Dublin to model the mechanism and develop a machine learning algorithm. The algorithm predicts which position on the ring the alkyl group will end up on based on an index of electron density. The team also partnered with AstraZeneca to run the reaction in a flow system on the gram scale.
Working on organic radical chemistry was “really at the border or beyond the comfort zone” for the group, Reisner says. The team was driven by a “fundamental belief that this reaction is really exciting and it needs to be explored,” and it was a great learning experience.
Chemical & Engineering News