Тел.: +7 (383) 330-67-71, факс: +7 (383) 330-80-56, E-mail: bic@catalysis.ru
630090, Россия, Новосибирск, пр-т Ак. Лаврентьева, 5
Тел.: +7 (383) 330-67-71, факс: +7 (383) 330-80-56, E-mail: bic@catalysis.ru
630090, Россия, Новосибирск, пр-т Ак. Лаврентьева, 5
Manganese edges into the realm of noble-metal photochemistry
Pairing manganese with an off-the-shelf ligand yields a low-cost complex that mimics precious-metal photocatalysts
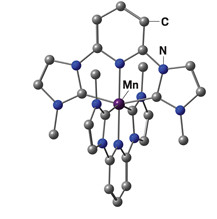 This manganese complex, with a record
excited-state lifetime of 190 ns, could one day be a low-cost alternative to precious-metal catalysts for light-driven reactions
This manganese complex, with a record
excited-state lifetime of 190 ns, could one day be a low-cost alternative to precious-metal catalysts for light-driven reactions
Precious metals such as ruthenium and iridium have long set the standard for photocatalysis because their complexes can absorb visible light and hold that energy for hundreds of nanoseconds—long enough to drive useful chemical reactions. But those metals are scarce and costly, which limits largescale uses such as solar fuel production and industrial photochemistry.
Now, researchers at Johannes Gutenberg University Mainz report a manganese complex that stays energized for 190 ns after absorbing light—a record for low-cost abundant metals (Nat. Commun. 2025, DOI: 10.1038/s41467-025-63225-4). The duration rivals what noble-metal catalysts can deliver and marks the first time a manganese system has reached the timescales needed for practical light-driven chemistry.
Katja Heinze, who led the work, says the breakthrough came from a deliberate pairing of the metal and a carbene-pyridine ligand. “Manga-nese binds this ligand more strongly than similar earth-abundant metals,” she explains. “That keeps the energy from leaking away too quickly, so the light-excited state lasts much longer.”
The ligand itself isn’t new—it was first made more than 2 decades ago simply to explore basic coordination chemistry. Kenneth Wärnmark’s group at Lund University later adapted the ligand for iron-based light absorbers, and that history inspired Heinze to test it with manganese. Wärnmark, who was not part of the new study, says Heinze’s choice to return to this first-generation ligand was crucial. “Second- and third-generation ligands are much stronger donors,” he explains, and “would have changed the way manganese handles energy, shutting down the long-lived excited state.”
Heinze adds that the choice of ligand was practical as well. Unlike more elaborate modern designs, this version is commercially available. “That opens the door to large-scale implementation,” she says.
This rational design didn’t come at the expense of performance. “Swapping iron for manganese stretched the lifetime from less than a trillionth of a second to 190 billionths,” Heinze says. “That window is long enough for the complex to interact with other molecules before the energy fades—something iron complexes still can’t achieve.” The team demonstrated this by successfully transferring an electron to benzophenone—a standard test molecule.
The complex also proved about 100 times as resistant to light-induced breakdown as ruthenium bipyridine complexes, a longtime benchmark for photocatalysts, underscoring its potential for durable, light-driven processes.
Both researchers emphasize that important work remains before the complex can serve as a practical catalyst. Heinze says the next step is to tweak the system so that the manganese complex can be regenerated after it reacts, without sacrificing its record-breaking energized state.
“If the world has to move on from fossil fuels, it will need large-scale production of solar fuels,” Wärnmark explains. “Noble metals simply aren’t abundant enough to support that, and that’s where such complex-es could one day make a difference.” Echoing Heinz, however, he adds that the manganese system so far works only once per molecule, not in a repeating catalytic cycle. “There is still a long way to go.”
Chemists finally know why palladium beats nickel at C–H activation
New study examines the precious metal’s ability to acidify bonds in alkanes
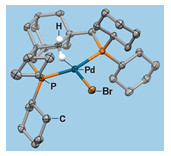 This X-ray structure of a model complex created by Demyan Prokopchuk’s lab illuminates how palladium (blue) interacts with hydrogen (white) to facilitate C–H activation.
This X-ray structure of a model complex created by Demyan Prokopchuk’s lab illuminates how palladium (blue) interacts with hydrogen (white) to facilitate C–H activation.
Reactions that sever typically unreactive carbon-hydrogen bonds, known as C-H activations, are a well-studied staple of organic chemis-try. So it’s a well-known fact that palladium-based catalysts tend to work better than ones based on palladium’s cheaper, possibly greener first-row counterpart, nickel.
But among the reams of scholarly papers about C–H activation methods and mechanisms,
“Really, nobody took a step back and asked, ‘How do we systematically even measure the C-H bond strength in this activation step?’ ” he says. He and his team at Rutgers University–Newark took it upon themselves to change that.
The plan was simple, as these things go: create model complexes—one nickel based, the other palladium based, but otherwise identical—in which the metal center coordinates to a carbon-hydrogen bond in a care-fully chosen alkane pincer ligand. The next step was to experimentally determine using nuclear magnetic resonance spectroscopy and acid-base equilibria how much each metal weakens, or activates, the bond.
Prokopchuk and his group published their initial results on the nickel complex in 2022 (J. Am. Chem. Soc. DOI: 10.1021/jacs.2c05667). Their investigation into the palladium complex came out on Sept. 9 (J. Am. Chem. Soc. 2025, DOI: 10.1021/jacs.5c07649).
Synthesizing the complexes was fairly straightforward, Prokopchuk says. By using X-ray crystallography, the researchers got a good “solid-state snapshot,” he says, of a key intermediate containing an agostic interaction, meaning the hydrogen atom is bound partly to carbon and partly to the metal center.
Measuring the acid-base equilibrium turned out to be harder for palladium than nickel because palladium had a tendency to reversibly form bimetallic dimers. It took a couple of years to figure out what was going on and how to disentangle the data to measure the proton transfer equilibrium the researchers were looking for.
“This is interesting, but also often complicated chemistry,” says Andreas Hansen of the University of Bonn, whose group handled the computa-tional aspects of the study. There was a lot for both the experimentalists and theorists to sink their teeth into in the project.
Ultimately, the researchers found that the palladium complex renders the C–H bond around 100,000 times as acidic (read: willing to part with H+) as it is in the nickel complex.
This not only gives quantitative weight to empirical observations of how palladium and nickel behave but may also help chemists figure out how to optimize their catalytic systems. For example, it suggests that nickel catalysts would benefit from being paired with stronger bases.
Tianning Diao, an organic chemist at New York University who studies C–H activation but was not involved with this work, describes the paper in an email to C&EN as “remarkable in many respects.” The synthesis and characterization of the palladium complex show “outstanding creativity in molecular design” and accompany a thorough mechanistic study that “provides long-sought experimental evidence for a hypothesis that has been speculated on” in the C–H activation community.
Prokopchuk says his team’s next steps are to continue with more head-to-head comparisons of other catalyst metals, such as cobalt, iridium, and rhodium.
Chemical & Engineering News