Тел.: +7 (383) 330-67-71, факс: +7 (383) 330-80-56, E-mail: bic@catalysis.ru
630090, Россия, Новосибирск, пр-т Ак. Лаврентьева, 5
Тел.: +7 (383) 330-67-71, факс: +7 (383) 330-80-56, E-mail: bic@catalysis.ru
630090, Россия, Новосибирск, пр-т Ак. Лаврентьева, 5
Bis-thiourea compound helps stereospecifically build β-glycoside sugar linkages
Assembling oligosaccharides and polysaccharides can be tricky business. The steric and electronic elements that come into play when hooking one sugar onto another makes the synthesis of these molecules unpredictable. Now, inspired by the enzymes that connect sugars to one another in cells, Eric N. Jacobsen and coworkers at Harvard University have developed a macrocyclic catalyst that predictably and reliably builds certain types of sugar linkages—known as β-glycosidic bonds—in a stereospecific manner (Science 2017, DOI: 10.1126/science.aal1875).
Jacobsen’s group was hoping to use its expertise in chiral catalysts to influence the stereochemical outcome of a substitution reaction of a glycosyl chloride to create oligosaccharides. But the team discovered that when using a macrocyclic bis-thiourea catalyst, both enantiomers of the catalyst led to the same stereochemical outcome in the product.
Instead of the catalyst controlling the stereochemistry of the reaction, the stereochemistry of the sugar controls the reaction, Jacobsen explains. The reaction takes place via an SN2 mechanism: The thiourea groups in the catalyst make hydrogen bonds to the chloride on the sugar to make it a better leaving group, while an amide side chain in the catalyst activates an alcohol to make it a better nucleophile. The team used the catalyst to assemble a wide variety of trans-1,2-, cis-1,2-, and 2-deoxy-β-glycosides.
“There’s no reliable, general way to attach any two sugars together in a predictable way,” Jacobsen says. “That’s key if we’re going to help advance the field of oligosaccharide synthesis, including automated methods, for biological applications.” This new catalyst, he says, is a step in that direction.
“This is a fascinating study that links concepts from numerous parts of chemistry to define a new approach to selective carbohydrate synthesis,” says Scott J. Miller, an expert in organic synthesis at Yale University. “Achieving catalyst control with nonenzymatic catalysts, through the use of the reported bis-thioureas, opens up new doors and leverages the physical organic chemistry of the catalytic mechanisms in powerful new ways.”
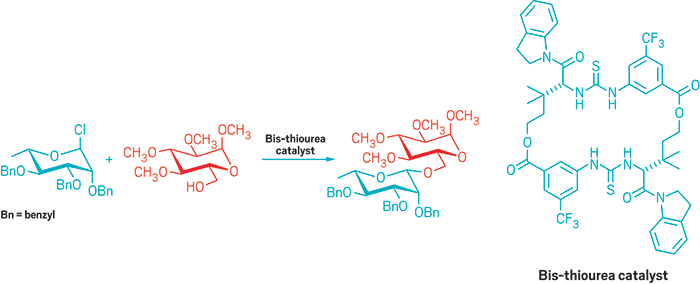
Carbohydrate connection: a macrocyclic bis-thiourea catalyst allows the Jacobsen group to create a ß-glycoside.
New method, dubbed MINFLUX, achieves nanometer resolution and fast analysis by combining the best of earlier imaging techniques
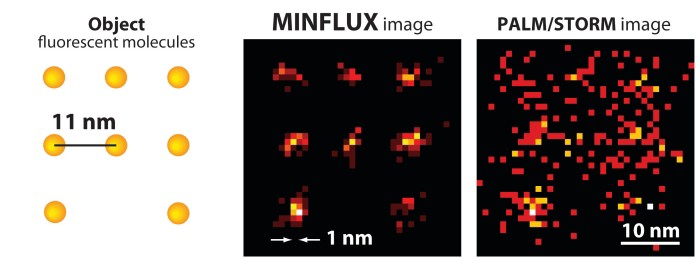
An array of fluorescent molecules (left) can be better resolved by MINFLUX (center) than by PALM/STORM (right).
Existing superresolution fluorescence microscopy methods such as STED and PALM have trouble imaging molecules with spatial resolution better than 20 to 40 nm. A new superresolution method called MINFLUX combines aspects of those earlier methods to resolve molecules just 6 nm apart with nanometer precision. The method is fast enough for short- and long-range tracking of individual molecules (Science 2016, DOI: 10.1126/science.aak9913).
STED and PALM (and the related technique STORM) distinguish densely packed features by allowing isolated molecules to fluoresce while those around them remain dark. The methods differ in how they determine the position of that fluorescence. In STED, a laser beam focused into a doughnut shape turns off fluorescence beneath the beam but not at the doughnut’s center. In PALM, fluorescent molecules are randomly switched on and off repeatedly so that only a few well-separated ones are activated at a time. The position of the activated molecules is determined with a camera.
“The strength of STED is that the doughnut always determines where the molecules are on and off and hence where the signal comes from,” says Stefan W. Hell of the Max Planck Institute for Biophysical Chemistry, who won the 2014 Nobel Prize in Chemistry for STED and leads the team that invented MINFLUX. On the other hand, “the strength of PALM is that you’reworking with individual molecules.” But the disadvantage of PALM is that those fluorescent molecules must emit many thousands of photons in order for scientists to get enough fluorescence intensity to determine their positions precisely.
In the new method, Hell, Francisco Balzarotti, also at the Max Planck Institute for Biophysical Chemistry, and coworkers usea doughnut-shaped laser beam for exciting fluorescence instead of turning it off. Because there’s no light intensity in the doughnut hole, a molecule located entirely within the hole won’t fluoresce. But a molecule that’s even slightly offset from the hole does fluoresce. How much it fluoresces depends on how far it is from the beam’s zero-intensity center. By scanning the doughnut in a defined pattern over the sample, the researchers can determine the precise position of a molecule from how its fluorescence emission changes.
“Because we can relate the position of the molecule to the ‘zero’ of the doughnut, many fewer photons are required to find out the position of the molecule than in PALM,” Hell says.
The team used a molecular assembly strategy called DNA origami to construct two arrays of fluorescent molecules at defined distances from one another: 11 nm in one array and 6 nm in the other. MINFLUX determined the molecules’ positions with a precision of 2.1 nm and 1.2 nm, respectively.
In addition to improved spatial resolution, the MINFLUX measurements are fast enough and require such low laser intensity that the researchers used the method for tracking individual molecules in live cells at multiple time scales.
“It’s a big deal that this method can be used for imaging but also for tracking at short and long time scales,” says Christy F. Landes, a superresolution expert at Rice University. “Not all methods are useful for these three very different imaging applications.”
Researchers create new reagents for a fluorinated version of copper-catalyzed azide-alkyne click chemistry
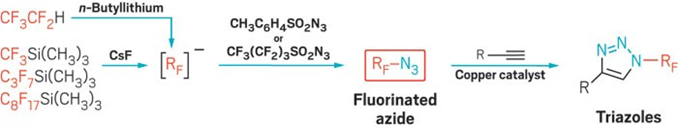
A two-step, one-pot reaction creates new fluorinated azides for copper-catalyzed fluorinated click chemistry.
With the help of new fluorinating reagents, a team led by Zsófia E. Blastik and Petr Beier of the Czech Academy of Sciences has devised a versatile fluorinated version of copper-catalyzed click chemistry. The approach overcomes some previous difficulties with preparing suitable fluorinated reagents for click reactions and opens the door to broader use of click chemistry, which has become invaluable to chemical biology and materials science.
The new chemistry hinges on the ability to make azidoperfluoroalkanes. Azidotrifluoromethane (CF3N3) has been known for some time, but its best reported synthesis starting from CF3I requires cumbersome handling of toxic and corrosive CF3NO, N2H4, and Cl2 gases—an approach that has limited CF3N3’s applications. Beier’s group alternatively attempted using electrophilic CF3I with sodium azide (NaN3) as a nucleophile, but the reaction didn’t work. Instead, the team found that CF3N3 and its previously unknown longer chain analogs can be prepared more conveniently from CF3Si(CH3)3 and related nucleophiles and sulfonyl azide electrophiles. In effect, the researchers flipped the polarity of the reaction’s constituents (Angew. Chem. Int. Ed. 2016, DOI: 10.1002/anie.201609715).
The reported azidoperfluoroalkanes undergo copper-catalyzed azide-alkyne cycloadditions, also known as click reactions, leading to N-perfluoroalkyl triazoles as underexplored building blocks, Beier says. “In fact, azidoperfluoroalkanes are more reactive in the click reaction with alkynes than nonfluorinated alkyl azides.”
“A reminder emerging out of this paper by Beier and colleagues is that, if you want to build a bond by a polar mechanism, there are always two options,” says Andrei K. Yudin of the University of Toronto, who focuses on the chemistry of heterocyclic compounds. “It is a good idea to reverse the polarity of components if one path is problematic. As a result, they have developed an efficient entry into azidoperfluoroalkanes.”
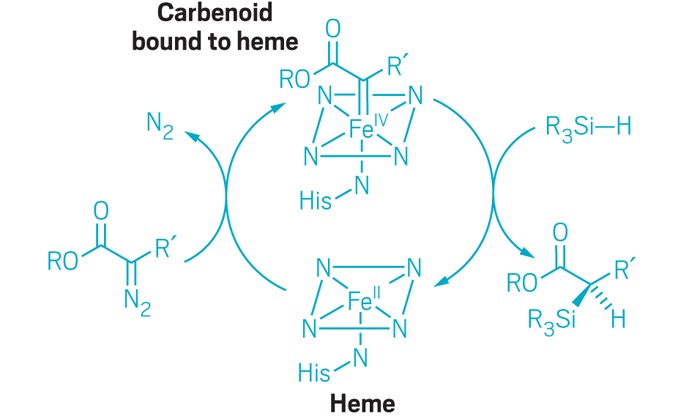
Bacterial cytochrome c demonstrates first example of biologically catalyzed organosilicon chemistry
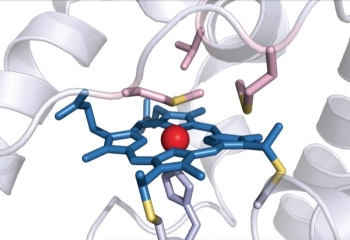 |
The heme group of R. marinus cytochrome c catalyzes carbenoid insertion into Si-H bonds. |
Silicon is the second most abundant element in Earth’s crust after oxygen, but carbon-silicon bonds are unheard of in nature: Neither biological organosilicon compounds nor biosynthetic pathways to create them have been identified. But when given the right starting materials, some heme proteins can stereospecifically form carbon-silicon bonds, report researchers from Caltech (Science 2016, DOI: 10.1126/science.aah6219).
“Nature’s iron heme chemistry just jumps on this opportunity because we provided it with the right precursors,” says Frances H. Arnold, who led the work with S. B. Jennifer Kan. “It’s a profound demonstration of how easily nature can innovate.”
 |
Three mutations of cytochrome c turn the protein into an enzyme that produces organosilicon compounds with greater than 99% enantiomeric excess. |
“This closes a crucial gap between biological and chemical catalysis,” writes Martin Oestreich of the Technical University of Berlin in a commentary accompanying the paper. “The impact is unforeseeable, but it seems that we are a big step closer to potentially facilitating industrially relevant reactions such as alkene hydrosilylation with biomolecules.”
Chemists have experimented for decades with forming chiral organosilicon compounds by carbenoid insertion into Si–H bonds, but the reactions typically require halogenated solvents and catalysts made of precious metals coordinated with chiral ligands. The catalysts also have low turnover—each catalyst complex survives fewer than 100 reactions before it inactivates.
Prior work in Arnold’s lab and elsewhere had demonstrated that heme proteins can catalyze nonnatural carbene transfer reactions through insertion into N–H and S–H bonds. In the new experiments, the researchers screened a panel of heme proteins to find ones that could catalyze insertion of ethyl 2-diazopropanoate into the Si–H bond of dimethyl(phenyl)silane. Cytochrome c from the bacterium Rhodothermus marinus, which is found in submarine hot springs in Iceland, catalyzed the reaction with 97% enantiomeric excess, although the catalytic turnover was still low.
Cytochrome c proteins normally don’t catalyze chemical reactions; instead they transfer electrons between biomolecules in cells. But that did not stop Kan, Arnold, and colleagues from pushing the R. marinus cytochrome c to improve its newfound ability to perform organosilicon catalysis.
The researchers used directed evolution to come up with a set of three mutations that together increase the new enzyme’s enantioselectivity to greater than 99% and turnover to greater than 1,500. One of the mutations involved a methionine residue that provides an axial ligand to the protein’s iron center. A change from methionine to aspartic acid possibly provided more substrate access for the formation of an iron-carbenoid intermediate.
The “evolved” cytochrome c performs stereoselective carbene insertion into Si–H bonds using a variety of silicon and diazo reagents, without competing cyclopropanation. And when given 4-(dimethylsilyl)aniline, which can accommodate insertion at both N–H and Si–H bonds, the enzyme formed the organosilicon product with 97% chemoselectivity. Arnold thinks that the partially exposed active site promotes substrate promiscuity but that there’s also some characteristic of the heme pocket that prefers silicon chemistry.
Chemical & Engineering News
ChemCatChem
Origin of the Improved Performance in Lanthanum-doped Silica-supported NiCatalysts
D. Baudouin, T. Margossian, U. Rodemerck, P.B. Webb, L. Veyre, F. Krumeich, J.-P. Candy, C. Thieuleux, C. Copére
DOI: 10.1002/cctc.201600582
Advanced Synthesis & Catalysis
Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions
H. Pellissier
DOI: 10.1002/adsc.201600462
Angewandte Chemie International Edition
Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and
Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry
J. Klankermayer, S. Wesselbaum, K. Beydoun, W. Leitner
DOI: 10.1002/anie.201507458
Chemistry – A European Journal
A One-Pot Synthesis of N-Aryl-2-Oxazolidinones and Cyclic
Urethanes by the Lewis Base Catalyzed Fixation of Carbon Dioxide into Anilines and Bromoalkanes
T.Niemi, J.E. Perea-Buceta, I. Fernández, O.-M. Hiltunen, V. Salo,S. Rautiainen, M.T. Räisänen, T. Repo
DOI: 10.1002/chem.201602338
ChemBioChem
Widening
the Product Profile of Carbon Dioxide Reduction by Vanadium Nitrogenase
J. G. Rebelein, Y. Hu, M.W. Ribbe
DOI: 10.1002/cbic.201500305
Chemistry – An Asian Journal
Silver(I)-Catalyzed
Three-Component Reaction of Propargylic Alcohols, Carbon Dioxide and Monohydric Alcohols: Thermodynamically Feasible Access to
β-Oxopropyl Carbonates
Zhi-Hua Zhou, Qing-Wen Song, Liang-Nian He et al
DOI: 10.1002/asia.201600600
ChemPlusChem
Electrocatalytic
Reduction of Carbon Dioxide with a Well-Defined PN3−Ru Pincer Complex
S. Min, S. Rasul, H. Li, D.C. Grills, K. Takanabe,L.-J. Li,K.-W. Huang
DOI: 10.1002/cplu.201500474
Advanced Materials Interfaces
Constructing
Ru/TiO2 Heteronanostructures Toward Enhanced Photocatalytic Water Splitting via a
RuO2/TiO2 Heterojunction and Ru/TiO2 Schottky
Junction
Q.Gu, Z. Gao, S. Yu, C. Xue
DOI: 10.1002/admi.201500631
Advanced Science
Visible and Near-Infrared Photothermal Catalyzed Hydrogenation of Gaseous
CO2 over Nanostructured Pd@Nb2O5
J.Jia, P.G. O'Brien, L. He, Q. Qiao, Teng Fei, L.M. Reyes, T.E. Burrow, Y. Dong, K. Liao, M. Varela, S.J. Pennycook, M. Hmadeh, A.S. Helmy, N.P. Kherani, D.D. Perovic, G.A. Ozin
DOI: 10.1002/advs.201600189
Energy
Technology
Enhancement
of the Efficiency and Selectivity for Carbon Dioxide
Electroreduction to Fuels on Tailored Copper Catalyst Architectures
M. Bevilacqua, J. Filippi, M. Folliero, A. Lavacchi,H.A. Miller, A. Marchionni, F. Vizza
DOI: 10.1002/ente.201600044
Macromolecular Chemistry and Physics
The
Synthesis of Poly(ethylene glycol) (PEG) Containing Polymers via
Step-Growth Click Coupling Reaction for CO2 Separation
B. N. Gacal, V.Filiz, V. Abetz
DOI: 10.1002/macp.201500381
Particle Systems Characterization
Progress
of Carbon Quantum Dots in Photocatalysis Applications
Z.Zhang, T. Zheng, X. Li, J. Xu, H. Zeng
DOI: 10.1002/ppsc.201500243